Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergAbstract
An understanding of evolution at the molecular level requires the simultaneous consideration of the 5 fundamental evolutionary processes: mutation, recombination, natural selection, genetic drift, and population dynamic effects. Experimental, comparative genomic, and population genetic work in C. elegans has greatly expanded our understanding of these core processes, as well as of C. elegans biology. This chapter presents a brief overview of some of the most salient features of molecular evolution elucidated by the C. elegans system.
Molecular evolution is the study of change-over-time in DNA sequence-based features, be they nucleotide variant frequencies in a population or divergence of loci between reproductively isolated lineages. This field has a rich theoretical history, and intertwines with classical and molecular population genetics and comparative genomics (Avise, 2004; Fisher, 1958; Gibson and Muse, 2002; Hartl and Clark, 2007; Li, 1997; Lynch, 2007). Facilitated by C. elegans' experimental tractability and the early publication of its genome sequence, this species has contributed greatly to our understanding of general processes of molecular evolution – and molecular evolutionary analyses have contributed greatly to our understanding of C. elegans biology. Like all descriptions of evolution, molecular evolution may be decomposed into 5 fundamental ingredients: mutation, recombination, natural selection, genetic drift, and population dynamic effects. In this brief overview to C. elegans molecular evolution, I will consider each of these features in turn to provide an introduction to the molecular organization and change-over-time of C. elegans genome. One broad review (Cutter et al., 2009), several targeted reviews (Coghlan et al., 2006; Thomas, 2008), and WormBook chapters (Natural variation and population genetics of Caenorhabditis elegans, Nematode genome evolution, The phylogenetic relationships of Caenorhabditis and other rhabditids and Gene duplications and genetic redundancy in C. elegans) provide complementary in-depth perspectives on aspects of C. elegans molecular evolution, in addition to the articles describing the genomes and genetic maps of C. elegans (Barnes et al., 1995; C. elegans Sequencing Consortium, 1998) and C. briggsae (Hillier et al., 2007; Stein et al., 2003).
Mutation provides the raw material for evolutionary change. Consequently, a comprehensive understanding of molecular evolution requires the characterization of mutational rates, mechanisms, biases, and heterogeneity. Accurate measures of mutational properties also are critical for molecular clock approaches to inferring the timing of historical evolutionary events that are recorded in patterns of polymorphism and divergence, such as coalescent times within populations, splitting times among populations, speciation events, and the timing of gene duplications (Coghlan and Wolfe, 2002; Cutter, 2008; Cutter et al., 2006; Lynch and Conery, 2000).
Among eukaryotes, C. elegans' mutation rates and mechanisms are among the most well-characterized, in large-part due to mutation accumulation (MA) experiments (Baer et al., 2007). Laboratory MA experiments that enable even moderately deleterious mutations to accrue in worm genomes over the course of dozens or hundreds of generations permit the indirect (Baer et al., 2006; Keightley and Bataillon, 2000; Keightley and Caballero, 1997; Vassilieva et al., 2000; Vassilieva and Lynch, 1999) or direct (Denver et al., 2009; Denver et al., 2004a; Denver et al., 2004b; Denver et al., 2000) inference of the average mutation rate across the genome. The most recent incarnation of this work, using whole-genome re-sequencing of several MA lines, indicates an average per-generation single-nucleotide mutation rate in the nuclear genome of 2.7 × 10−9 (Denver et al., 2009), which is roughly 3-times lower than a previous direct-sequencing analysis of MA lines (Denver et al., 2004b). Small indels in the nuclear genome are thought to arise at an average rate per generation that is comparable to the single-nucleotide rate (Denver et al., 2004b), although it is now unclear whether this result holds, in light of the most recent point-mutation rate estimates. Denver et al. (2004b) estimated that an average of roughly 1 deleterious mutation arises in the diploid genome per generation, which corroborates previous work that suggested that phenotypic estimates of the genomic deleterious mutation rate were much too low (Davies et al., 1999). Different DNA repair pathways influence mutation rates to varying degrees, with a particularly strong role of mismatch repair (Denver et al., 2006). In the mitochondrial genome, the mutation rate is inferred to average 9.7 × 10−8 mutations per site per generation (Denver et al., 2000). Curiously, mitochondrial nucleotide mutational biases differ strikingly among yeast, fly, mouse and worm (Montooth and Rand, 2008).
Short tandem repeat loci (STRs, or, microsatellites) are popular molecular markers (Barrière and Félix, 2007; Haber et al., 2005; Sivasundar and Hey, 2003), because typically they have high mutation rates that generate relatively high heterozygosity and because most are thought to be selectively neutral. STRs mutate by a mechanism distinct from single nucleotide changes, such that slippage of the replication machinery generates mutant alleles that differ in the number of repeats by one or more units. Studies of dysfunctional repair mutants (Degtyareva et al., 2002) and MA experiments in C. elegans (Denver et al., 2004a; Phillips et al., 2009; Seyfert et al., 2008) both have informed mutational dynamics of STRs. Longer STRs (loci with more repeats, regardless of the length of the repeat unit) mutate faster, and also typically exhibit higher population genetic variation (Phillips et al., 2009). Nearly 30% of observed mutations involve multi-step changes, and longer STRs may have a greater fraction of multi-step changes – i.e., mutation is not consistent with a strict stepwise-mutation model, SMM (Ohta and Kimura, 1973). Because most mutational studies focus on long STRs (which mutate faster and are easier to study), yet most STRs in the worm genome are quite short, the incidence of single-step changes for typical STRs may actually be substantially more than 70% and more closely approximate the SMM.
Copy number variants originate by mutation from duplications, insertions, deletions, and the special case of transposable element activity. Such mutations are now recognized to contribute to substantial differences among wild isolates (Maydan et al., 2007) and between species (Stein et al., 2003). Most duplications occur intra-chromosomally, generally are less than 2kb in length (Cavalcanti et al., 2003; Katju and Lynch, 2003; Semple and Wolfe, 1999; Vergara et al., 2009), and arise at a rate much higher than in other taxa (Cutter et al., 2009; Lynch and Conery, 2000). More detailed discussion of duplication in the worm genome is provided elsewhere (Cutter et al., 2009; The putative chemoreceptor families of C. elegans, Gene duplications and genetic redundancy in C. elegans). Rearrangements form another important class of mutation, which, in C. elegans, occur primarily within rather than between chromosomes and at a rate substantially higher than in other taxonomic groups (Coghlan and Wolfe, 2002; Hillier et al., 2007; Stein et al., 2003; Vergara et al., 2009). Thus, Caenorhabditis displays higher mutation rates for several classes of mutations (single-nucleotide, duplication, rearrangement) relative to other taxa.
Recombination generates new combinations of alleles from heterozygous individuals, and can facilitate natural selection by reducing interference between linked alleles that are subject to selection simultaneously. Consequently, recombination should be important for the effective elimination of deleterious alleles (via negative directional, purifying selection) and for the fixation of beneficial alleles (via positive directional selection). Because genetically effective recombination requires heterozygosity, and inbreeding by self-fertilization and biparental inbreeding destroys heterozygosity, understanding the role of recombination for highly selfing C. elegans in nature is a major issue for molecular evolution (Phillips, 2006).
Crossover rates vary along C. elegans chromosomes in a predictable manner (see Karyotype, ploidy and gene dosage): the central half of each chromosome has distinctly low rates of crossover, the arms flanking the chromosome centers experience high rates of crossover, and the chromosome tips have virtually no crossover (Barnes et al., 1995; Rockman and Kruglyak, 2009). This pattern is most pronounced on the autosomes, but still detectable on the X sex-chromosome. C. briggsae's chromosomes exhibit a comparable pattern of crossover (Cutter and Choi, unpublished data; Hillier et al., 2007), so this may represent the ancestral and universal chromosomal pattern of crossover in Caenorhabditis – but confirmation in gonochoristic species like C. remanei is necessary. Chromosomes experience essentially complete interference, such that just a single crossover occurs per chromosome in meiosis (excepting the hemizygous X in males) (Zetka, 2009), and these crossovers do not appear to occur at recombination “hotspots” as in yeast and mammals.
The chromosomal distribution of crossover rates in C. elegans has important implications for nucleotide polymorphism and divergence. Notably, single-nucleotide differences and indel polymorphisms among strains are more common in regions of high recombination (Cutter and Payseur, 2003; Koch et al., 2000; Maydan et al., 2007). It has been proposed that this might reflect higher rates of mutation in regions of high recombination and/or that natural selection more dramatically impacts regions of low recombination (due to longer spans of linkage associated with selected sites), resulting in depressed levels of polymorphism. Indeed, divergence relative to C. briggsae at silent sites is greater in high recombination regions (Cutter and Payseur, 2003), as are rates of chromosomal rearrangement (Hillier et al., 2007; Stein et al., 2003), consistent with a higher mutation rate in regions experiencing elevated crossover rates. However, whole-genome sequencing of MA lines identified no significant heterogeneity along the length of chromosomes with respect to mutation rate, based on ∼400 detected mutations (Denver et al., 2009). Natural selection could potentially shape chromosomal distributions of genetic variation in C. elegans as well (Cutter and Payseur, 2003). “Background selection” (Charlesworth et al., 1993; Hudson and Kaplan, 1995) against deleterious mutations was proposed as a plausible selective explanation for low polymorphism in regions that experience little recombination, though genetic hitchhiking due to positive directional selection also might contribute to the pattern (Andolfatto, 2001; Smith and Haigh, 1974; Wiehe and Stephan, 1993). The proteins encoded in chromosomal centers (i.e. low crossover rate regions) tend to be more highly conserved across taxa (C. elegans Sequencing Consortium, 1998; Parkinson et al., 2004), although the rate of amino acid substitution (KA) does not strongly correlate with crossover rate once the substitution rate at synonymous sites (KS) is properly accounted for (including adjustment for selection on synonymous sites). Analysis of this latter point should be revisited with species comparisons more closely related than C. elegans and C. briggsae, between which divergence at synonymous sites is saturated.
Gene conversion is an important process shaping linkage disequilibrium at very small scales within genomes (Andolfatto and Nordborg, 1998). Gene conversion as a mechanism of recombination remains fairly poorly understood generally, and in C. elegans in particular. Specific examples of allelic gene conversion have been reported (Moerman and Baillie, 1979; Rattray and Rose, 1988; Rose and Baillie, 1980), as well as evidence of gene conversion among gene duplicates or multi-gene families in the genome (Katju et al., 2008; Semple and Wolfe, 1999), and from population polymorphism data (in C. remanei) (Cutter, 2008). Gene conversion is now widely implemented for experimental purposes in C. elegans (Frøkjaer-Jensen et al., 2008; Robert et al., 2008), which holds promise for furthering our understanding of the process of gene conversion and its implications for molecular evolution. However, no genome-wide contrast of rates of crossover versus non-crossover (i.e. gene conversion) recombination have been conducted, as in yeast (Mancera et al., 2008).
No conclusive evidence of mitochondrial recombination has been reported for C. elegans, despite support for mitochondrial recombination in other nematodes (Lunt and Hyman, 1997; Piganeau et al., 2004) and a potential example in C. briggsae (Howe and Denver, 2008).
Genetically effective recombination occurs only rarely in nature, such that large blocks of chromosomes experience linkage disequilibrium among wild isolates (Barrière and Félix, 2005; Cutter, 2006; Haber et al., 2005; Rockman and Kruglyak, 2009). Linkage disequilibrium (LD) takes an average of 3.3Mb to decay by half, and significant LD occurs between loci on different chromosomes. Very low rates of outcrossing in nature (on the order of 10−3 to 10−4 per generation) are thought to be the primary cause of this LD, although complex population structure may also contribute. Despite the strong LD across the genome, clearly some recombination has occurred among multi-locus genotypes, meaning that there is no single bifurcating gene-tree that describes the population history of wild isolates of C. elegans. It should also be noted that microsatellite heterozygosity yields higher estimates of outcrossing rates, and there might be important variation among local populations in outcrossing rates (Sivasundar and Hey, 2005), perhaps a consequence of harsh or variable local conditions (Morran et al., 2009; Morran et al., 2009). However, selection may generally disfavor recombinant genotypes (Barrière and Félix, 2007; Dolgin et al., 2007). Extreme levels of self-fertilization and lack of effective recombination, such as observed in C. elegans, can have dire consequences for the long-term persistence of populations, in the absence of compensatory mutations (Loewe and Cutter, 2008).
A principle aim of molecular evolutionists is to identify and characterize the targets of adaptive evolution across the genome. However, to infer the action of selection, one must first refute the possibility that neutral evolutionary forces (i.e. the 4 evolutionary processes other than natural selection) could have caused the observed patterns. This is the reason that molecular evolutionists and molecular population geneticists so often are interested in the evolutionary dynamics of loci that are expected to be unaffected by selection; understanding the evolution of such neutral sites provides a null model against which to test for evidence of selection.
Population genetic polymorphism and inter-specific divergence are the two main tools for inferring selection at the molecular level. Unfortunately, the low diversity coupled with high LD due to the complex population processes in C. elegans (selfing, structured populations) render polymorphism as largely uninformative for localizing the gene targets of selection in this species. Consequently, most work has focused on analyses of divergence. When assessing nucleotide divergence between species, it is common practice to quantify the number of substitutions per basepair in the site class under consideration (e.g. replacement sites versus synonymous sites within coding regions) and to account for the potential for multiple mutational hits at a given site to have occurred. This is done with the various ways of computing KA (or, dN; the number of replacement substitutions per replacement site) and KS (or, dS; the number of synonymous substitutions per synonymous site) statistics (Yang and Bielawski, 2000). Dividing KA by KS provides a metric that relates the rate of change at amino acid-changing sites relative to the rate expected for putatively neutrally evolving synonymous sites. In other words, differences in KS are meant to reflect differences among loci in the underlying local rate of mutation that could, in itself, give rise to differences in rates of protein divergence. However, because synonymous sites for highly expressed genes have not been selectively neutral in the lineages leading to species of Caenorhabditis, it is further necessary to correct KS values for the effects of selection on codon usage (Cutter, 2008).
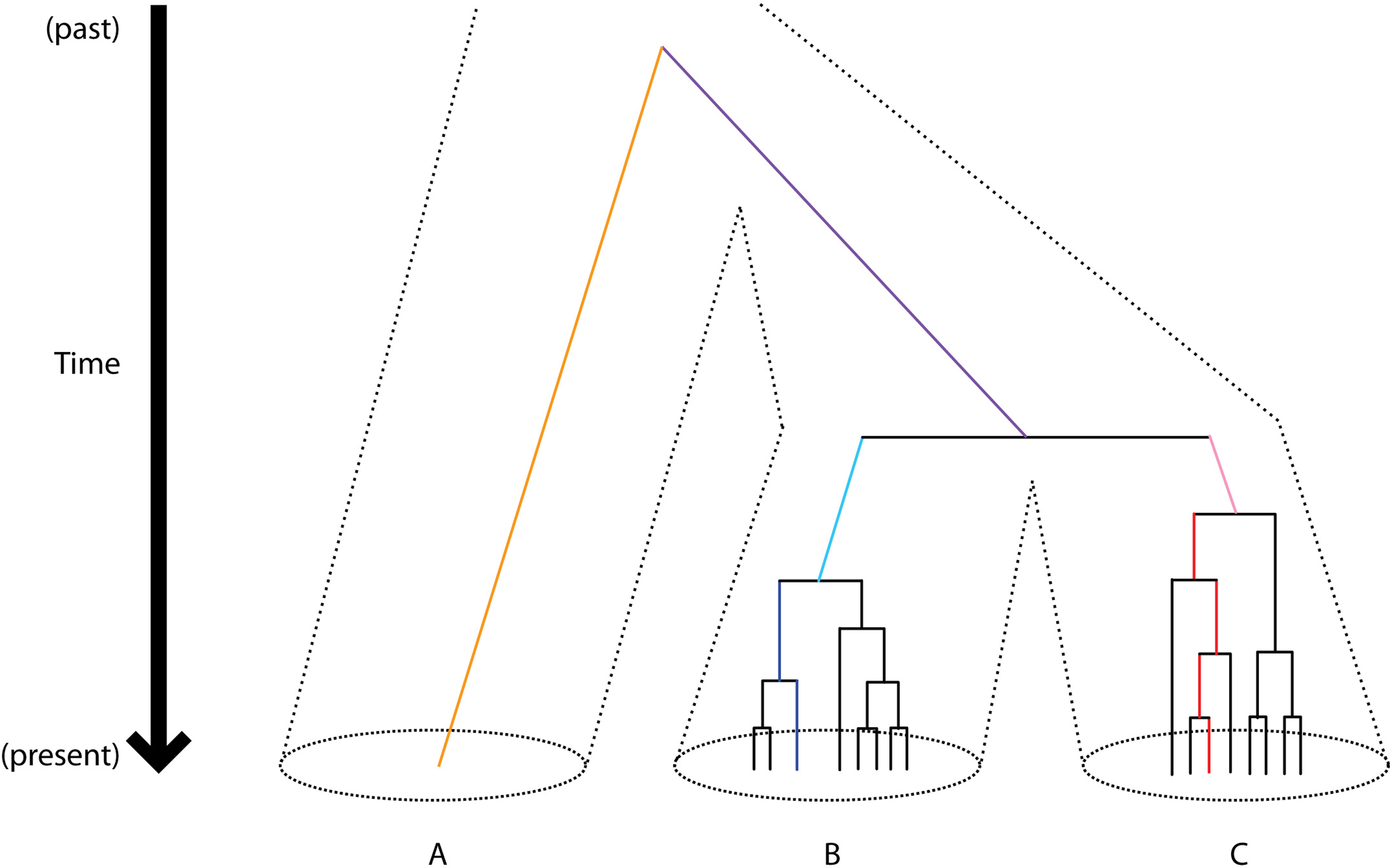
It is assumed implicitly in most divergence calculations that within-species polymorphism is negligible relative to fixed differences between species, which is a reasonable assumption for comparisons of known Caenorhabditis species (which are highly divergent at the molecular level), but may not be reasonable in other taxa (e.g. some Drosophila species pairs, human-chimpanzee) and may become an important issue as new, closely-related species of Caenorhabditis are discovered. Note also that some methods of computing divergence are pairwise, whereas others use a phylogenetic tree to generate lineage-specific rate estimates separately for each branch in the tree (Figure 1). Such lineage-specific measures of divergence require that the phylogenetic relationships among species are well-defined (Kiontke et al., 2007; The phylogenetic relationships of Caenorhabditis and other rhabditids); as genome sequences become available for additional Caenorhabditis and other nematodes, new developments in multi-locus species-tree inference should prove useful in delineating the tempo of speciation (Degnan and Rosenberg, 2009).
|
Figure 1. Diagrammatic representation of polymorphism and divergence. Dotted lines delimit independently evolving lineages (species); solid lines trace the genealogy of an ortholog between species. Polymorphism at a locus is shown within species B and C, as represented by a genealogy in each species that coalesces into the single common ancestor of the extant polymorphism within that lineage. Only a single representative copy of the locus is represented for species A. The expected coalescent time for extant polymorphism is 4Ne generations. Divergence between distantly-related species typically involves comparison of a single representative sequence for a locus from each species. For example, if the lengths of branches are proportional to the number of mutations that have accumulated along them, then divergence between A and B would be the sum of the lengths of the dark and light blue, purple, and orange branches. Using the colored copies of the locus as representatives from each of these 3 species, one could calculate lineage-specific rates for species B and C. Lineage-specific divergence for C (orange) would be equivalent to summing the lengths of the red and pink branches, if the position of their common ancestry could be inferred accurately. Differences in mutation rates and/or generation times can result in variable lineage-specific branch-lengths.
The dominant form of natural selection in the genome, of course, is purifying selection (also termed negative directional selection), which acts to conserve sequence features and, in coding sequences, yields KA/KS < 1 (Stein et al., 2003). For example, purifying selection exerts particularly potent effects on genes that confer sterility phenotypes when knocked-down by RNAi (Cutter et al., 2003). Also, adult-expressed genes tend to evolve more rapidly than those expressed predominantly in larvae, which is consistent with stronger purifying selection on pre-reproductive stages, as predicted by some theoretical models of aging (Cutter and Ward, 2005). Significant portions of non-coding sequence also are subject to purifying selection in Caenorhabditis (Kent and Zahler, 2000; Shabalina and Kondrashov, 1999; Webb et al., 2002), which has been applied to successfully identify conserved regulatory elements (Coghlan et al., 2006). However, many functionally important sequences may evolve rapidly, and therefore be missed in genomic surveys of conserved elements.
Detecting positive selection can be a difficult task, particularly in C. elegans, because many of the most powerful methods are not accessible or robust in this system due to the high sequence divergence between species in combination with C. elegans' complex demographic history (Kreitman, 2000). Comparative genomic analysis with newly identified species with closer phylogenetic positions to known species will help rectify this problem in the future. Nevertheless, a variety of intriguing general molecular evolutionary patterns resulting from natural selection reveal themselves under close inspection.
Within multi-gene families, it is possible to identify genes with an excess of replacement-site substitutions relative to synonymous-site substitutions (i.e. KA/KS > 1), which is indicative of repeated positive directional selection on peptide sequence (Yang and Bielawski, 2000). Specifically, the D subfamily of the ATP-binding cassette family of proteins shows evidence of such adaptive evolution in its history (Zhao et al., 2007), as do some members of the SRZ subfamily of putative chemoreceptors (Thomas et al., 2005), some lysozyme genes (Schulenburg and Boehnisch, 2008), and some antimicrobial peptides (Pujol et al., 2008). Transcription factors also tend to evolve more rapidly than other gene classes (Haerty et al., 2008; Jovelin, 2009), although strictly speaking the data for Caenorhabditis make it difficult to rule out relaxed selection as a possible cause of elevated rates of replacement-site divergence. Similarly, sperm-related genes tend to have more rapid rates of protein evolution than other types of genes, and also experience disproportionate gene loss and duplication (Artieri et al., 2008; Cutter and Ward, 2005); this could be a signature of sexual selection in the obligately outbreeding ancestors of C. elegans. As gene networks and genetic pathways become better-elucidated, molecular evolution analysis in the frameworks of systems biology and evo-devo may provide interesting insights (Jovelin, 2009; Jovelin et al., 2009; Zou et al., 2008).
Stochastic change in allele frequency describes the process of genetic drift (Hartl and Clark, 2007), and this mechanism of evolutionary change is important for alleles that are very rare and/or subject to very weak natural selection. Drift at a given locus is most powerful when populations are composed of few breeding individuals; the effect of genetic drift on an allele is inversely proportional to Nes (selection coefficient s). Ne, the effective population size, is a unifying concept in evolution that defines the size of populations not in terms of census size but in terms of the rate of genetic change due to drift (Charlesworth, 2009). Since the origin of the highly-selfing form of hermaphroditism that C. elegans now experiences in nature, the role of genetic drift in molecular evolution will have been increased dramatically throughout C. elegans' genome (a decrease in Ne relative to obligately outcrossing ancestors). Phrased another way, the relative importance of selection has been relaxed in the C. elegans lineage since the origin of extreme selfing.
The evolution of biased codon usage provides an exceptional example of the interplay between mutation, selection, and drift. These three forces operate with similar magnitudes of effect to determine the evolutionary fate of alternative synonymous codons (Bulmer, 1991; Duret, 2002). Mutational pressures and natural selection clearly both play important roles in the biased usage of synonymous codons in Caenorhabditis (Cutter et al., 2008; Duret, 2000; Duret and Mouchiroud, 1999; Stenico et al., 1994), and nematodes generally (Cutter et al., 2006). However, the drastic reduction in the effective number of breeding individuals in C. elegans, following the origin of a highly selfing lifestyle from obligately outbreeding ancestors (Cho et al., 2004; Kiontke et al., 2004), will have raised the relative importance of genetic drift in mediating molecular evolution. This yields the prediction that adaptive codon usage bias should be less prevalent in the C. elegans genome compared to its relatives, with the extent of codon bias decay being proportional to the time over which such relaxed selection has elapsed. Based on this logic, and the observation that codon bias differs only subtly between C. elegans and other species in the genus, Cutter et al. (2008) concluded that the extreme form of selfing seen today in C. elegans must have originated in the not-too-distant past.
The activity of transposable elements (TEs) most typically exerts detrimental consequences on organismal fitness (Begin and Schoen, 2007), but the ability of natural selection to eliminate such deleterious mutational events depends on the relative role of genetic drift. Again, the drastically reduced population size of C. elegans relative to its ancestor is predicted to have relaxed selection against the elimination of TE insertions. Population frequencies for 32 Tc1 insertion sites are consistent with relaxed selection in C. elegans (i.e. a dominant role for genetic drift in contemporary natural populations), in stark contrast to evidence of purifying selection against mTcre1 elements in C. remanei (Dolgin et al., 2008).
The Bergerac strains of C. elegans (e.g. BO = RW7000) were isolated from nature by Nigon, and exhibit an exceptionally high load of Tc1 elements (>300) in their genomes (Emmons et al., 1983; Hodgkin and Doniach, 1997; Liao et al., 1983). Several other C. elegans strains at the Caenorhabditis Genetics Center similarly have high loads of Tc1 (Hodgkin and Doniach, 1997), although all of these appear to reflect spurious mislabeling and/or lab crosses with Bergerac during their history in the lab (Rockman and Kruglyak, 2009). It is tempting to speculate that relaxed selection on transposon activity in C. elegans enabled the proliferation of Tc1 in Bergerac, which presumably eventually led to its extinction, as other strains with high genomic loads of Tc1 have not been isolated in recent years of extensive wild sampling effort. However, the isolation of Bergerac in the 1940's followed by decades of laboratory culture (Hodgkin and Doniach, 1997) raises the distinct possibility that the high abundance of Tc1 elements in its genome occurred after its isolation from nature.
Relaxed selection on chemoreceptor genes similarly might be responsible for the segregation of premature stop codon alleles being common in nature (Stewart et al., 2005). The evolution of genes underlying traits associated with outcrossing and sexual selection, such as copulatory plugs (Palopoli et al., 2008), also likely currently are governed largely by genetic drift.
In addition to the important impacts of mutation, recombination, selection, and drift on the evolution of inherited molecules, the details of population demography (population subdivision, migration, changes in size of the breeding population, mode of reproduction) also influence the ultimate evolutionary fate of alleles: loss or fixation. Molecular population genetics has been instrumental in tracing the ancestry of humans across the globe (Conrad et al., 2006; Rosenberg et al., 2002), and similar approaches have yielded generous insights into the dynamics of contemporary populations for C. elegans and its relatives (Natural variation and population genetics of Caenorhabditis elegans, Cutter et al., 2009; Fitch, 2005; Phillips, 2006).
C. elegans in nature exhibits strong evidence of subdivided population structure, with many subpopulations that display little-if-any isolation by distance (Barrière and Félix, 2005; Cutter, 2006; Haber et al., 2005; Sivasundar and Hey, 2005). This implies migration across both local and global scales, perhaps mediated by humans or other animal vectors. Migration and/or colonization of the globe may occur sufficiently frequently or recently that no obvious patterns of local adaptation are now evident for C. elegans, as inferred from molecular population genetic analysis and lack of associations between phenotype, genotype, and geography (Hodgkin and Doniach, 1997; Sivasundar and Hey, 2003). Some local subpopulations appear to be stable over time, while others go extinct and are recolonized (Barrière and Félix, 2007). These observations make metapopulation dynamic models and many-demes models of coalescent genealogical history relevant to understanding its genetic variation (Cutter, 2006; Pannell, 2003; Pannell and Charlesworth, 1999; Wakeley, 1999; Wakeley and Aliacar, 2001). However, it remains unclear precisely how to define a “subpopulation” for these nematodes in an ecological sense: do the worms in an ephemeral rotting apple comprise a subpopulation? Or is the soil under the apple tree, present for years or decades, home to a subpopulation? Or the grounds of the orchard in which the apple tree resides? Integrating C. elegans ecological context – which presently remains poorly known – with evolutionary analysis should prove helpful in elucidating the dynamics of population processes.
While molecular approaches find no clear support for global population expansion, and only limited support for population contraction (Sivasundar and Hey, 2003), the power to detect changes in effective population size in C. elegans very recent past is constrained by the complex population structure. Nevertheless, the substantially lower levels of genetic diversity (~20-fold) relative to obligately outcrossing species imply that it has experienced a strong genetic bottleneck since the origin of highly selfing hermaphroditism, possibly due to both demographic and selective causes (Cutter et al., 2009; Graustein et al., 2002).
Comparisons of molecular variation for multiple populations across species of Caenorhabditis that exhibit similar (e.g., C. briggsae, C. sp. 11) or different (e.g., C. remanei, C. brenneri, C. sp. 5) modes of reproduction to C. elegans also will help illuminate population dynamic processes. For example, despite the presence of selfing hermaphroditism and a global distribution like C. elegans, C. briggsae exhibits strong patterns of phylogeography (reviewed in Cutter et al., 2009; Cutter et al., 2006; Cutter et al., 2010; Howe and Denver, 2008). Current evidence also points to much greater levels of genetic variation in obligately outbreeding female-male species (Barriere et al., 2009; reviewed in Cutter et al., 2009; Jovelin, 2009; Jovelin et al., 2009). These observations point to important differences in the demography of these species that confer unique opportunities to exploit the peculiarities of a given species to inform the processes of molecular evolution and the biology of Caenorhabditis.
Study of C. elegans has greatly informed understanding of the fundamental forces of evolution operating at the molecular level, and the application of evolutionary theory to C. elegans genome has benefited understanding of this exquisite organism. Mutation perhaps is the best characterized aspect of molecular evolution in C. elegans. However, the great wealth of knowledge on mutation is matched by a relatively nascent understanding of the generality of mutational characteristics across species – and across greater phylogenetic depths. Despite the advances to molecular evolution made possible by research on C. elegans, it is now clear that investigation of other species of Caenorhabditis will provide complimentary and novel insights that are not possible by studying C. elegans alone (Haag et al., 2007; Kammenga et al., 2008). In particular, the roles of ecology and adaptation in shaping genetic variation, genomic heterogeneity, phenotypic variation, and phylogeography in the lineage that gave rise to C. elegans and its relatives will benefit profoundly from studies of obligately outcrossing species and from further comparative analysis of the repeated evolution of the selfing hermaphroditic lifestyle. The feasibility of resequencing entire Caenorhabditis genomes and transcriptomes with next-generation technologies promises advances in molecular evolutionary understanding with this group. For example, we should all anticipate rapid genome data acquisition for newly discovered species with closer phylogenetic ties to other known taxa (e.g., C. sp. 9), population genomics analysis of outcrossing species (e.g., C. remanei) to infer targets of natural selection from patterns of population polymorphism, and large-scale functional assays in non-elegans species. The scope of such advances, however, currently is limited by taxon sampling in Caenorhabditis to species that are rather distantly-related – collection and identification of new species in the genus is paramount to long-term progress in characterizing molecular evolutionary phenomena with Caenorhabditis.
A.D.C. is supported by a Canada Research Chair in Evolutionary Genomics and the Natural Sciences and Engineering Research Council of Canada. Thanks to R. Jovelin and two anonymous reviewers for their comments on the manuscript.
Andolfatto, P. (2001). Adaptive hitchhiking effects on genome variability. Curr. Opin. Genet. Dev. 11, 635-641. Abstract
Andolfatto, P., and Nordborg, M. (1998). The effect of gene conversion on intralocus associations. Genetics 148, 1397-1399. Abstract
Artieri, C. G., Haerty, W., Gupta, B. P., and Singh, R. S. (2008). Sexual selection and maintenance of sex: evidence from comparisons of rates of genomic accumulation of mutations and divergence of sex-related genes in sexual and hermaphroditic species of Caenorhabditis. Mol. Biol. Evol. 25, 972-979. Abstract Article
Avise, J. C. (2004). Molecular Markers, Natural History, and Evolution, 2nd ed. (Sunderland, MA, Sinauer).
Baer, C. F., Miyamoto, M. M., and Denver, D. R. (2007). Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat. Rev. Genet. 8, 619-631. Abstract Article
Baer, C. F., Phillips, N., Ostrow, D., Avalos, A., Blanton, D., Boggs, A., Keller, T., Levy, L., and Mezerhane, E. (2006). Cumulative effects of spontaneous mutations for fitness in Caenorhabditis: role of genotype, environment and stress. Genetics 174, 1387-1395. Abstract Article
Barnes, T. M., Kohara, Y., Coulson, A., and Hekimi, S. (1995). Meiotic recombination, noncoding DNA and genomic organization in Caenorhabditis elegans. Genetics 141, 159-179. Abstract
Barrière, A., and Félix, M.-A. (2005). High local genetic diversity and low outcrossing rate in Caenorhabditis elegans natural populations. Curr. Biol. 15, 1176-1184. Abstract Article
Barrière, A., and Félix, M.-A. (2005). Natural variation and population genetics of Caenorhabditis elegans. In WormBook, The C. elegans Research Community, ed., http://www.wormbook.org.
Barrière, A., and Félix, M.-A. (2007). Temporal dynamics and linkage disequilibrium in natural Caenorhabditis elegans populations. Genetics 176, 999-1011. Abstract Article
Barrière, A., Yang, S. P., Pekarek, E., Thomas, C. G., Haag, E. S., and Ruvinsky, I. (2009). Detecting heterozygosity in shotgun genome assemblies: Lessons from obligately outcrossing nematodes. Genome Res. 19, 470-480. Abstract Article
Begin, M., and Schoen, D. J. (2007). Transposable elements, mutational correlations, and population divergence in Caenorhabditis elegans. Evolution 61, 1062-1070. Abstract Article
Bulmer, M. (1991). The selection-mutation-drift theory of synonymous codon usage. Genetics 129, 897-907. Abstract
C. elegans Sequencing Consortium (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012-2018. Abstract
Cavalcanti, A. R., Ferreira, R., Gu, Z., and Li, W. H. (2003). Patterns of gene duplication in Saccharomyces cerevisiae and Caenorhabditis elegans. J. Mol. Evol. 56, 28-37. Abstract Article
Charlesworth, B. (2009). Fundamental concepts in genetics: Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 10, 195-205. Abstract Article
Charlesworth, B., Morgan, M. T., and Charlesworth, D. (1993). The effect of deleterious mutations on neutral molecular variation. Genetics 134, 1289-1303. Abstract
Cho, S., Jin, S. W., Cohen, A., and Ellis, R. E. (2004). A phylogeny of Caenorhabditis reveals frequent loss of introns during nematode evolution. Genome Res. 14, 1207-1220. Abstract Article
Coghlan, A. (September 7, 2005). Nematode genome evolution. In WormBook, The C. elegans Research Community, ed., http://www.wormbook.org.
Coghlan, A., Stajich, J. E., and Harris, T. W. (2006). Comparative genomics in C. elegans, C. briggsae, and other Caenorhabditis species. In C. elegans: Methods and Applications, K. Strange, ed. (Totowa, NJ, Humana Press Inc.), pp. 13-29.
Coghlan, A., and Wolfe, K. H. (2002). Fourfold faster rate of genome rearrangement in nematodes than in Drosophila. Genome Res. 12, 857-867. Abstract Article
Conrad, D. F., Jakobsson, M., Coop, G., Wen, X. Q., Wall, J. D., Rosenberg, N. A., and Pritchard, J. K. (2006). A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet. 38, 1251-1260. Abstract Article
Cutter, A. D. (2006). Nucleotide polymorphism and linkage disequilibrium in wild populations of the partial selfer Caenorhabditis elegans. Genetics 172, 171-184. Abstract Article
Cutter, A. D. (2008). Divergence times in Caenorhabditis and Drosophila inferred from direct estimates of the neutral mutation rate. Mol. Biol. Evol. 25, 778-786. Abstract Article
Cutter, A. D. (2008). Multilocus patterns of polymorphism and selection across the X-chromosome of Caenorhabditis remanei. Genetics 178, 1661-1672. Abstract Article
Cutter, A. D., Dey, A., and Murray, R. L. (2009). Evolution of the Caenorhabditis elegans genome. Mol. Biol. Evol. 26, 1199-1234. Abstract Article
Cutter, A. D., Félix, M.-A., Barriere, A., and Charlesworth, D. (2006). Patterns of nucleotide polymorphism distinguish temperate and tropical wild isolates of Caenorhabditis briggsae. Genetics 173, 2021-2031. Abstract Article
Cutter, A. D., and Payseur, B. A. (2003). Selection at linked sites in the partial selfer Caenorhabditis elegans. Mol. Biol. Evol. 20, 665-673. Abstract Article
Cutter, A. D., Payseur, B. A., Salcedo, T., Estes, A. M., Good, J. M., Wood, E., Hartl, T., Maughan, H., Strempel, J., Wang, B., et al. (2003). Molecular correlates of genes exhibiting RNAi phenotypes in Caenorhabditis elegans. Genome Res. 13, 2651-2657. Abstract Article
Cutter, A. D., and Ward, S. (2005). Sexual and temporal dynamics of molecular evolution in C. elegans development. Mol. Biol. Evol. 22, 178-188. Abstract Article
Cutter, A. D., Wasmuth, J., and Blaxter, M. L. (2006). The evolution of biased codon and amino acid usage in nematode genomes. Mol. Biol. Evol. 23, 2303-2315. Abstract Article
Cutter, A. D., Wasmuth, J. D., and Washington, N. L. (2008). Patterns of molecular evolution in Caenorhabditis preclude ancient origins of selfing. Genetics 178, 2093-2104. Abstract Article
Cutter, A. D., Yan, W., Tsvetkov, N., Sunil, S., and Félix, M. A. (2010). Molecular population genetics and phenotypic sensitivity to ethanol for a globally diverse sample of the nematode Caenorhabditis briggsae. Mol. Ecol.
Davies, E. K., Peters, A. D., and Keightley, P. D. (1999). High frequency of cryptic deleterious mutations in Caenorhabditis elegans. Science 285, 1748-1751. Abstract
Degnan, J. H., and Rosenberg, N. A. (2009). Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332-340. Abstract Article
Degtyareva, N. P., Greenwell, P., Hofmann, E. R., Hengartner, M. O., Zhang, L., Culotti, J. G., and Petes, T. D. (2002). Caenorhabditis elegans DNA mismatch repair gene msh-2 is required for microsatellite stability and maintenance of genome integrity. Proc. Natl. Acad. Sci. USA 99, 2158-2163. Abstract Article
Denver, D. R., Dolan, P. C., Wilhelm, L. J., Sung, W., Lucas-Lledo, J. I., Howe, D. K., Lewis, S. C., Okamoto, K., Thomas, W. K., Lynch, M., and Baer, C. F. (2009). A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc. Natl. Acad. of Sci. USA 106, 16310-16324. Abstract Article
Denver, D. R., Feinberg, S., Steding, C., Durbin, M., and Lynch, M. (2006). The relative roles of three DNA repair pathways in preventing Caenorhabditis elegans mutation accumulation. Genetics 174, 57-65. Abstract Article
Denver, D. R., Morris, K., Kewalramani, A., Harris, K. E., Chow, A., Estes, S., Lynch, M., and Thomas, W. K. (2004a). Abundance, distribution, and mutation rates of homopolymeric nucleotide runs in the genome of Caenorhabditis elegans. J. Mol. Evol. 58, 584-595. Abstract Article
Denver, D. R., Morris, K., Lynch, M., and Thomas, W. K. (2004b). High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430, 679-682. Abstract Article
Denver, D. R., Morris, K., Lynch, M., Vassilieva, L. L., and Thomas, W. K. (2000). High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science 289, 2342-2344. Abstract
Dolgin, E. S., Charlesworth, B., Baird, S. E., and Cutter, A. D. (2007). Inbreeding and outbreeding depression in Caenorhabditis nematodes. Evolution 61, 1339-1352. Abstract Article
Dolgin, E. S., Charlesworth, B., and Cutter, A. D. (2008). Population frequencies of transposable elements in selfing and outcrossing Caenorhabditis nematodes. Genet. Res. 90, 317-329. Abstract Article
Duret, L. (2000). tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet. 16, 287-289. Abstract
Duret, L. (2002). Evolution of synonymous codon usage in metazoans. Curr. Opin. Genet. Dev. 12, 640-649. Abstract
Duret, L., and Mouchiroud, D. (1999). Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, Arabidopsis. Proc. Natl. Acad. Sci. USA 96, 4482-4487. Abstract
Emmons, S. W., Yesner, L., Ruan, K. S., and Katzenberg, D. (1983). Evidence for a transposon in Caenorhabditis elegans. Cell 32, 55-65. Abstract
Fitch, D. H. A. (2005). Evolution: An ecological context for C. elegans. Curr. Biol. 15, R655-R658. Abstract Article
Frøkjaer-Jensen, C., Davis, M. W., Hopkins, C. E., Newman, B. J., Thummel, J. M., Olesen, S. P., Grunnet, M., and Jorgensen, E. M. (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375-1383. Abstract Article
Graustein, A., Gaspar, J. M., Walters, J. R., and Palopoli, M. F. (2002). Levels of DNA polymorphism vary with mating system in the nematode genus Caenorhabditis. Genetics 161, 99-107. Abstract
Haag, E. S., Chamberlin, H., Coghlan, A., Fitch, D. H., Peters, A. D., and Schulenburg, H. (2007). Caenorhabditis evolution: if they all look alike, you aren't looking hard enough. Trends Genet. 23, 101-104. Abstract Article
Haber, M., Schungel, M., Putz, A., Muller, S., Hasert, B., and Schulenburg, H. (2005). Evolutionary history of Caenorhabditis elegans inferred from microsatellites: Evidence for spatial and temporal genetic differentiation and the occurrence of outbreeding. Mol. Biol. Evol. 22, 160-173. Abstract Article
Haerty, W., Artieri, C., Khezri, N., Singh, R. S., and Gupta, B. P. (2008). Comparative analysis of function and interaction of transcription factors in nematodes: extensive conservation of orthology coupled to rapid sequence evolution. BMC Genomics 9, 399. Abstract Article
Hartl, D. L., and Clark, A. G. (2007). Principles of Population Genetics, 4th ed. (Sunderland, MA, Sinauer Associates).
Hillier, L. W., Miller, R. D., Baird, S. E., Chinwalla, A., Fulton, L. A., Koboldt, D. C., and Waterston, R. H. (2007). Comparison of C. elegans and C. briggsae genome sequences reveals extensive conservation of chromosome organization and synteny. PLoS Biology 5, e167. Abstract Article
Hodgkin, J. (June 25, 2005). Karyotype, ploidy and gene dosage. In WormBook, The C. elegans Research Community, ed., http://www.wormbook.org.
Hodgkin, J., and Doniach, T. (1997). Natural variation and copulatory plug formation in Caenorhabditis elegans. Genetics 146, 149-164. Abstract
Howe, D. K., and Denver, D. R. (2008). Muller's ratchet and compensatory mutation in Caenorhabditis briggsae mitochondrial genome evolution. BMC Evol. Biol. 8, 62. Abstract Article
Hudson, R. R., and Kaplan, N. L. (1995). Deleterious background selection with recombination. Genetics 141, 1605-1617. Abstract
Jovelin, R. (2009). Rapid sequence evolution of transcription factors controlling neuron differentiation in Caenorhabditis. Mol. Biol. Evol. 26, 2373-2386. Abstract Article
Jovelin, R., Dunham, J. P., Sung, F. S., and Phillips, P. C. (2009). High nucleotide divergence in developmental regulatory genes contrasts with the structural elements of olfactory pathways in Caenorhabditis. Genetics 181, 1387-1397. Abstract Article
Kammenga, J. E., Phillips, P. C., De Bono, M., and Doroszuk, A. (2008). Beyond induced mutants: using worms to study natural variation in genetic pathways. Trends Genet. 24, 178-185. Abstract Article
Katju, V., LaBeau, E. M., Lipinski, K. J., and Bergthorsson, U. (2008). Sex change by gene conversion in a Caenorhabditis elegans fog-2 mutant. Genetics 180, 669-672. Abstract Article
Katju, V., and Lynch, M. (2003). The structure and early evolution of recently arisen gene duplicates in the Caenorhabditis elegans genome. Genetics 165, 1793-1803. Abstract
Keightley, P. D., and Bataillon, T. M. (2000). Multigeneration maximum-likelihood analysis applied to mutation-accumulation experiments in Caenorhabditis elegans. Genetics 154, 1193-1201. Abstract
Keightley, P. D., and Caballero, A. (1997). Genomic mutation rates for lifetime reproductive output and lifespan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 94, 3823-3827. Abstract
Kent, W. J., and Zahler, A. M. (2000). Conservation, regulation, synteny, and introns in a large-scale C. briggsae - C. elegans genomic alignment. Genome Res. 10, 1115-1125. Abstract
Kiontke, K., Barriére, A., Kolotuev, I., Podbilewicz, B., Sommer, R., Fitch, D. H., and Félix, M. A. (2007). Trends, stasis, and drift in the evolution of nematode vulva development. Curr. Biol. 17, 1925-1937. Abstract Article
Kiontke, K., and Fitch, D. H. A. (August 11, 2005). The phylogenetic relationships of Caenorhabditis and other rhabditids. In WormBook, The C. elegans Research Community, ed., http://www.wormbook.org.
Kiontke, K., Gavin, N. P., Raynes, Y., Roehrig, C., Piano, F., and Fitch, D. H. A. (2004). Caenorhabditis phylogeny predicts convergence of hermaphroditism and extensive intron loss. Proc. Natl. Acad. Sci. USA 101, 9003-9008. Abstract Article
Koch, R., van Luenen, H. G. A. M., van der Horst, M., Thijssen, K. L., and Plasterk, R. H. A. (2000). Single nucleotide polymorphisms in wild isolates of Caenorhabditis elegans. Genome Res. 10, 1690-1696. Abstract
Kreitman, M. (2000). Methods to detect selection in populations with applications to the human. Annual Review of Genomics and Human Genetics 1, 539-559. Abstract Article
Liao, L. W., Rosenzweig, B., and Hirsh, D. (1983). Analysis of a transposable element in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 80, 3585-3589. Abstract
Loewe, L., and Cutter, A. D. (2008). On the potential for extinction by Muller's ratchet in Caenorhabditis elegans. BMC Evol. Biol. 8, 125. Abstract Article
Lunt, D. H., and Hyman, B. C. (1997). Animal mitochondrial DNA recombination. Nature 387, 247. Abstract Article
Lynch, M., and Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151-1155. Abstract
Mancera, E., Bourgon, R., Brozzi, A., Huber, W., and Steinmetz, L. M. (2008). High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454, 479-485. Abstract Article
Maydan, J. S., Flibotte, S., Edgley, M. L., Lau, J., Selzer, R. R., Richmond, T. A., Pofahl, N. J., Thomas, J. H., and Moerman, D. G. (2007). Efficient high-resolution deletion discovery in Caenorhabditis elegans by array comparative genomic hybridization. Genome Res. 17, 337-347. Abstract Article
Moerman, D. G., and Baillie, D. L. (1979). Genetic organization in Caenorhabditis elegans: fine-structure analysis of the unc-22 gene. Genetics 91, 95-103. Abstract
Montooth, K. L., and Rand, D. M. (2008). The spectrum of mitochondrial mutation differs across species. PLoS Biology 6, 1634-1637. Abstract Article
Morran, L. T., Cappy, B. J., Anderson, J. L., and Phillips, P. C. (2009). Sexual partners for the stressed: facultative outcrossing in the self-fertilizing nematode Caenorhabditis elegans. Evolution 63, 1473-1482. Abstract Article
Morran, L. T., Parmenter, M. D., and Phillips, P. C. (2009). Mutation load and rapid adaptation favour outcrossing over self-fertilization. Nature 462, 350-352. Abstract Article
Ohta, T., and Kimura, M. (1973). Model of mutation appropriate to estimate number of electrophoretically detectable alleles in a finite population. Genet. Res. 22, 201-204. Abstract
Palopoli, M. F., Rockman, M. V., Tinmaung, A., Ramsay, C., Curwen, S., Aduna, A., Laurita, J., and Kruglyak, L. (2008). Molecular basis of the copulatory plug polymorphism in Caenorhabditis elegans. Nature 454, 1019-1022. Abstract Article
Pannell, J. R. (2003). Coalescence in a metapopulation with recurrent local extinction and recolonization. Evolution 57, 949-961. Abstract
Pannell, J. R., and Charlesworth, B. (1999). Neutral genetic diversity in a metapopulation with recurrent local extinction and recolonization. Evolution 53, 664-676.
Parkinson, J., Mitreva, M., Whitton, C., Thomson, M., Daub, J., Martin, J., Schmid, R., Hall, N., Barrell, B., Waterston, R. H., et al. (2004). A transcriptomic analysis of the phylum Nematoda. Nat. Genet. 36, 1259-1267. Abstract Article
Phillips, N., Salomon, M., Custer, A., Ostrow, D., and Baer, C. F. (2009). Spontaneous mutational and standing genetic (co)variation at dinucleotide microsatellites in Caenorhabditis briggsae and Caenorhabditis elegans. Mol. Biol. Evol. 26, 659-669. Abstract Article
Piganeau, G., Gardner, M., and Eyre-Walker, A. (2004). A broad survey of recombination in animal mitochondria. Mol. Biol. Evol. 21, 2319-2325. Abstract Article
Pujol, N., Zugasti, O., Wong, D., Couillault, C., Kurz, C. L., Schulenburg, H., and Ewbank, J. J. (2008). Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathogens 4, e1000105. Abstract Article
Rattray, B., and Rose, A. M. (1988). Increased intragenic recombination and non-disjunction in the Rec-1 strain of Caenorhabditis elegans. Genet. Res. 51, 89-93.
Robert, V. J., Davis, M. W., Jorgensen, E. M., and Bessereau, J. L. (2008). Gene conversion and end-joining-repair double-strand breaks in the Caenorhabditis elegans germline. Genetics 180, 673-679. Abstract Article
Robertson, H. M., and Thomas, J. H. (January 06, 2006). The putative chemoreceptor families of C. elegans. In WormBook, The C. elegans Research Community, ed., http://www.wormbook.org.
Rockman, M. V., and Kruglyak, L. (2009). Recombinational landscape and population genomics of C. elegans. PLoS Genetics 5, e1000419. Abstract Article
Rose, A. M., and Baillie, D. L. (1980). Genetic organization of the region around UNC-15 (I), a gene affecting paramyosin in Caenorhabditis elegans. Genetics 96, 639-648. Abstract
Rosenberg, N. A., Pritchard, J. K., Weber, J. L., Cann, H. M., Kidd, K. K., Zhivotovsky, L. A., and Feldman, M. W. (2002). Genetic structure of human populations. Science 298, 2381-2385. Abstract Article
Schulenburg, H., and Boehnisch, C. (2008). Diversification and adaptive sequence evolution of Caenorhabditis lysozymes (Nematoda: Rhabditidae). BMC Evol. Biol. 8, 114. Abstract Article
Semple, C., and Wolfe, K. H. (1999). Gene duplication and gene conversion in the Caenorhabditis elegans genome. J. Mol. Evol. 48, 555-564. Abstract
Seyfert, A. L., Cristescu, M. E., Frisse, L., Schaack, S., Thomas, W. K., and Lynch, M. (2008). The rate and spectrum of microsatellite mutation in Caenorhabditis elegans and Daphnia pulex. Genetics 178, 2113-2121. Abstract Article
Shabalina, S. A., and Kondrashov, A. S. (1999). Pattern of selective constraint in C. elegans and C. briggsae genomes. Genet. Res. 74, 23-30. Abstract
Sivasundar, A., and Hey, J. (2003). Population genetics of Caenorhabditis elegans: the paradox of low polymorphism in a widespread species. Genetics 163. Abstract
Sivasundar, A., and Hey, J. (2005). Sampling from natural populations using RNAi reveals high outcrossing and population structure in Caenorhabditis elegans. Curr. Biol. 15, 1598-1602. Abstract Article
Smith, J. M., and Haigh, J. (1974). Hitch-hiking effect of a favorable gene. Genet. Res. 23, 23-35. Abstract
Stein, L. D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M. R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al. (2003). The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biology 1, 166-192. Abstract Article
Stenico, M., Lloyd, A. T., and Sharp, P. M. (1994). Codon usage in Caenorhabditis elegans: delineation of translational selection and mutational biases. Nucleic Acids Res. 22, 2437-2446. Abstract
Stewart, M. R., Clark, N. L., Merrihew, G., Galloway, E. M., and Thomas, J. H. (2005). High genetic diversity in the chemoreceptor superfamily of Caenorhabditis elegans. Genetics 169, 1985-1996. Abstract Article
Thomas, J. H. (2008). Genome evolution in Caenorhabditis. Brief. Func. Genom. Proteom. 7, 211-216. Abstract Article
Thomas, J. H., Kelley, J. L., Robertson, H. M., Ly, K., and Swanson, W. J. (2005). Adaptive evolution in the SRZ chemoreceptor families of Caenorhabditis elegans and Caenorhabditis briggsae. Proc. Natl. Acad. Sci. USA 102, 4476-4481. Abstract Article
Vassilieva, L. L., Hook, A. M., and Lynch, M. (2000). The fitness effects of spontaneous mutations in Caenorhabditis elegans. Evolution 54, 1234-1246. Abstract
Vassilieva, L. L., and Lynch, M. (1999). The rate of spontaneous mutation for life-history traits in Caenorhabditis elegans. Genetics 151, 119-129. Abstract
Vergara, I., Mah, A., Huang, J., Tarailo-Graovac, M., Johnsen, R., Baillie, D., and Chen, N. (2009). Polymorphic segmental duplication in the nematode Caenorhabditis elegans. BMC Genomics 10, 329. Abstract Article
Wakeley, J. (1999). Nonequilibrium migration in human history. Genetics 153, 1863-1871. Abstract
Wakeley, J., and Aliacar, N. (2001). Gene genealogies in a metapopulation. Genetics 159, 893-905. Abstract
Webb, C. T., Shabalina, S. A., Ogurtsov, A. Y., and Kondrashov, A. S. (2002). Analysis of similarity within 142 pairs of orthologous intergenic regions of Caenorhabditis elegans and Caenorhabditis briggsae. Nucleic Acids Res. 30, 1233-1239. Abstract
Wiehe, T. H. E., and Stephan, W. (1993). Analysis of a genetic hitchhiking model, and its application to DNA polymorphism data from Drosophila melanogaster. Mol. Biol. Evol. 10, 842-854. Abstract
Woollard, A. (June 25, 2005). Gene duplications and genetic redundancy in C. elegans. In WormBook, The C. elegans Research Community, ed., (http://www.wormbook.org).
Yang, Z., and Bielawski, J. P. (2000). Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496-503. Abstract
Zetka, M. (2009). Homologue pairing, recombination and segregation in Caenorhabditis elegans. Genome Dynamics 5, 43-55. Abstract Article
*Edited by David H. A. Fitch. Last revised December 3, 2009. Published March 5, 2010. This chapter should be cited as: Cutter, A. D. Molecular evolution inferences from the C. elegans genome (March 5, 2010), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.149.1, http://www.wormbook.org.
Copyright: © 2010 Asher D. Cutter. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: asher.cutter@utoronto.ca
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.