Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
The soil nematode Caenorhabditis briggsae is an attractive model system for studying evolution of both animal development and behavior. Being a close relative of C. elegans, C. briggsae is frequently used in comparative studies to infer species-specific function of the orthologous genes and also for studying the dynamics of chromosome evolution. The genome sequence of C. briggsae is valuable in reverse genetics and genome-wide comparative studies. This review discusses resources and tools, which are currently available, to facilitate study of C. briggsae in order to unravel mechanisms of gene function that confer morphological and behavioral diversity.
C. briggsae is a natural companion to C. elegans for research because of the similarities in morphology, hermaphroditic life style and ease of cultivation. The two species diverged from a common ancestor roughly 100 million years ago (see Section 2.4; Stein et al., 2003) yet appear almost identical in morphology and behavior. The phylogenetic relationships of Caenorhabditis nematodes reveals that C. briggsae and C. elegans are not sibling species and that C. remanei, a gonochoristic species, is the closest relative of C. briggsae (see The phylogenetic relationships of Caenorhabditis and other rhabditids). The seemingly identical morphology of the two nematodes permits straightforward interpretation of results involving conserved genes and pathways. The majority of the currently available genetic and reverse genetic tools in C. elegans can be used in C. briggsae with no major modification. These together with its whole genome sequence make C. briggsae an ideal model for comparative studies.
The whole genome sequencing project (Stein et al., 2003) revealed that the genomes of C. briggsae and C. elegans have much in common (Summarized in Table 1). For example, both worms have the same number of chromosomes (six chromosomes each), similar genome size, and similar numbers of protein coding and non-protein coding genes. Further analysis demonstrated that about 62% of the protein coding genes in C. briggsae have orthologs in C. elegans. Nevertheless, many interesting species-specific features including species-specific genes exist, which serve as the foundation for comparative analysis. In the following subsections, we will describe the C. briggsae genome and compare it with the C. elegans genome. Data described here are all available at WormBase (Chen et al., 2005).
A hybrid approach combining whole-genome assembly with whole-genome shotgun sequencing (WGS) was adopted to sequence the C. briggsae genome (Stein et al., 2003). The C. briggsae strain AF16 was targeted for physical map construction (Marra et al., 1997) and whole genome sequencing. A total number of 2.068 million reads, representing an approximately 10-fold coverage of the entire genome, were assembled into 5,341 contigs and linked together into 889 gapped supercontigs containing 105.6 Mbp of DNA sequence. This assembly (named cb25.agp8) consists of 463 sequence superconfigs placed into 142 FPCs and covers 102,431,873 bp and is publicly available at the WormBase FTP site. This assembly is estimated to cover 98% of the entire C. briggsae genome with a sequencing accuracy of approximately 99.98%. Therefore the C. briggsae genome size is approximately 104 Mbp, a size similar to but slightly larger than that of the C. elegans genome (102 Mbp, according to WormBase WS160). Recently, the construction of a genetic map using single nucleotide polymorphic (SNP) markers has positioned 100 Mb along the six chromosomes and refined the sequence map (Hillier et al., 2005).
Table 1. Comparison between genomes of C. briggsae and C. elegans (primarily based on Stein et al. (2003))
| Species | C. briggsae | C. elegans | |
|---|---|---|---|
| Genome size | 104 Mb | 102 Mb | |
| Sequencing approach | Combined physical mapping and WGS | Entirely clone-based | |
| Reads | 2.068 | - | |
| Most recent assembly | Cb25.agp8 | WormBase WS160 | |
| Assembly | 142 FPCs | 6 chromosomes | |
| Coming assembly | 6 chromosomes | - | |
| Repetitive elements content | 22.4% | 16.5% | |
| Protein-coding genes* | Number of genes | 19,507 | 20,621 |
| Exons | 114,339 | 125,702 | |
| Introns | 94,832 | 105,081 | |
| Median length | 1.90 kbp | 1.83 kbp | |
| Summed length | 55.7 Mbp | 55.6 Mbp | |
| Gene density | 5.4 kbp per gene | 4.9 kbp per gene | |
| Non-coding genes | tRNA | 777 (181) | 609 (210) |
| 5S rRNA | 7 | 15 | |
| 5.8S rRNA | 0 | 1 | |
| 18S rRNA | 0 (3) | 2 (1) | |
| 26S rRNA | 0 (7) | 1 (1) | |
| SRP | 4 | 5 | |
| U3 snoRNA | 4 | 6 | |
| U1 | 11 | 12 | |
| U2 | 15 | 20 | |
| U4 | 5 | 5 | |
| U5 | 10 | 15 | |
| U6 | 40 | 23 | |
| miRNA | 70 | 105 | |
| RNAaseP | 1 | 1 | |
| SL1 | 0 | 0 | |
| SL2 | 18 | 18 | |
| Notes: tRNA pseudogene predictions and other fragmentary matches are indicated in parentheses. rRNA genes and SL1 were found in tandemly duplicated arrays that were largely excluded from the genomic assembly. | |||
| *Protein-coding genes are “hybrid” genes that were predicted using four gene prediction programs (Genefinder, Fgenesh, Twinscan, and Ensembl pipeline). | |||
An interesting difference between the genomes of C. briggsae and C. elegans, which was revealed by comparative genomic analysis, is that the difference in the genome size is mainly due to the larger non-genic repetitive DNA content in C. briggsae's genome, which accounts for 22.4% (or 23.3 Mbp) of the C. briggsae genome compared to 16.5% (or 16.5 Mbp) of the C. elegans genome (see Table 1). Notably, a single C. briggsae repeat family, Cb000047, is present more than 20,000 times and covers 4.7 Mbp of the C. briggsae genome, while it is present only a few hundred times in the C. elegans genome. Despite the striking similarities between the genomes and coding sequences of C. briggsae and C. elegans as summarized in Table 1, only a few orthologous repetitive elements were identified between these two sister species, suggesting that essentially all extant dispersed repeat elements postdate the divergence of these two species.
Before a detailed description of the comparative analysis between C. briggsae and C. elegans protein-coding genes are made, some caveats should be pointed out. First, while the C. briggsae genome is still at a draft stage, the C. elegans genome sequencing is complete. Second, while the C. briggsae gene set was generated entirely by computational gene finding, the C. elegans gene set was generated by computational gene finding, followed by extensive manual curation with various experimental evidences including ESTs, OSTs, and full length cDNAs. Third, while only a small number of laboratories use C. briggsae as a major experimental organism, a significantly larger number of laboratories use C. elegans, which not only provides more data for accurately curating C. elegans gene modes, but also creates a strong pressure to establish a high-quality C. elegans gene set. In fact, the C. briggsae gene set (the "hybrid" gene set) remained the same for three years following the publication of the C. briggsae genome analysis paper (Stein et al., 2003) until recently when WormBase started to curate C. briggsae genes (WormBase Consortium, personal communication). In the meantime, the C. elegans gene set has been improved and updated in WormBase in each of its tri-weekly releases.
As shown in Table 1, there are similar numbers of protein-coding genes in the genomes of C. briggsae and C. elegans. The unspliced lengths of protein-coding genes and the total length of the protein-coding genes, including the length of their introns, are roughly the same between the two species, further suggesting that the larger size of the C. briggsae genome is not contributed by the number or size of the protein-coding genes. The C. briggsae genome has contributed to the improved predictions of C. elegans protein-coding genes. Notably, C. briggsae gene prediction has helped to predict 1,275 new C. elegans genes (Stein et al., 2003). So far, about 300 genes of these have been confirmed by OSTs of the ORFeome project. Despite the similarity of the number of genes shared by the two species, C. briggsae has about 800 genes that have not be found in C. elegans, while C. elegans has 1,061 genes that have not been found in C. briggsae (Stein et al., 2003).
One way C. briggsae and C. elegans orthologs were identified was by searching for C. briggsae/C. elegans pairs that were each other's best BLASTP (Altschul et al., 1997) match in the opposite species. Synteny was subsequently used as a criterion to search for more orthologous pairs. These combined approaches produced altogether 12,155 ortholog pairs, or 62% of the C. briggsae gene set or 65% of the C. elegans gene set (Stein et al., 2003). After the publication of the initial C. briggsae genome analysis paper, a recently developed program InParanoid (O'Brien et al., 2005) was applied to identify orthologs between C. briggsae and C. elegans. The InParanoid database (April 2005 release), using a recent C. elegans gene set, lists 12,858 C. briggsae and C. elegans ortholog pairs. Therefore the numbers of orthologs obtained by these two different approaches agree well with each other. However, the number of identified orthologs will change over time as the genome assembly and gene annotation, especially for C. briggsae, improve.
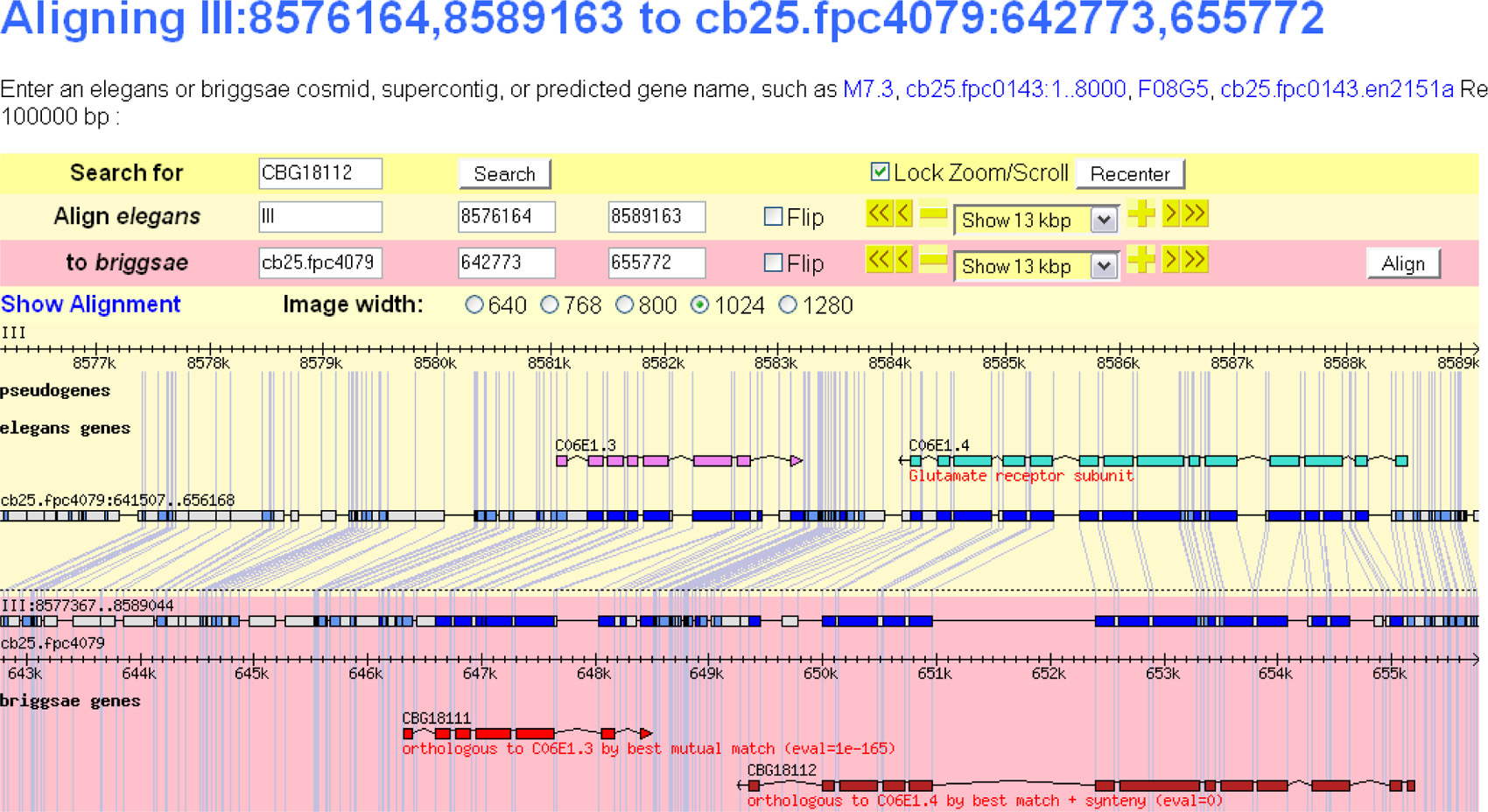
Currently the C. elegans ortholog of a C. briggsae gene is displayed in the Homology section on its gene page in WormBase and vice versa. Users can follow the link to the gene page of the C. elegans ortholog of a C. briggsae gene. Right next to this ortholog link, there is another link termed “syntenic alignment”, which directs users to a corresponding “Synteny Browser” page (see Figure 1). The Synteny Browser provides a comparative view of two genomic regions, a C. elegans genomic region that contains the C. elegan ortholog of the C. briggsae gene, a C. briggsae genomic region that is syntenic to the C. elegans region. The C. briggsae genomic region contains the C. briggsae ortholog of the C. elegans gene (see Figure 1). Users can browse along the genomic sequences by modifying the genomic coordinates.
|
Figure 1. WormBase synteny browser. A comparative view of syntenic regions of two genomes-C. elegans and C. briggsae genomes-is displayed in the browser. Also displayed are gene models contained within these genomic regions in both genomes and alignment details. Users can browse along genomic sequences by clicking on the Zoom/Scroll errors or modifying the genomic coordinates.
Before the C. briggsae genome sequencing project, the divergence of C. elegans and C. briggsae had been estimated to be 20–120 MYA (million years ago; Coghlan and Wolfe, 2002; Heschl and Baillie, 1990; Kennedy et al., 1993; Lee et al., 1992; Prasad and Baillie, 1989). The C. briggsae genome sequencing project helped to refine the divergence date to 80–110 MYA (Stein et al., 2003). The new estimation, based on 338 sets of orthologs, is more precise because of the large gene sample size and improved C. briggsae gene predictions and ortholog assignments.
There are more introns per gene in C. elegans than in C. briggsae (see Table 1). Comparative analysis, by aligning orthologous protein-coding genes between C. briggsae and C. elegans, revealed 6,579 species-specific introns among the 60,775 introns in the orthologous pairs of protein-coding genes. Roughly two-thirds (4,379) are C. elegans-specific, while one-third (2,200) are C. briggsae-specific, which suggest rates of intron gains or losses of about 0.5 per gene in the 80–110 million years since the split between C. briggsae and C. elegans. This rate is dramatically higher than that of mouse and human, which is 0.01 intron gains or losses per gene in 75 million years. Active intron gains and losses have also been observed by comparing many smaller gene sets in C. elegans and C. briggsae, for example, the chemosensory gene families (Robertson, 1998; Robertson, 2000; Robertson, 2001), a sets of five genes (four CPEB genes and fog-3; Cho et al., 2004), and another set of five genes (SSU rDNA, LSU rDNA, part of RNAP2, par-6, and pkc-3; Kiontke et al., 2004). These studies demonstrated dramatically higher rates of intron gain and loss in C. briggsae and C. elegans compared to that in mammals, and furthermore, significantly higher rates of intron loss compared to intron gain, which happen only rarely. Recently, a study identified 81 introns gained in C. elegans and 41 in C. briggsae (Coghlan and Wolfe, 2004). Among these 122 new introns, twenty-eight show significant DNA sequence similarity to other introns in the same genomes, including three that are similar to other introns in the same gene, suggesting that some of the introns were gained via reverse splicing of preexisting intron.
Whole genome sequence alignment of C. briggsae and C. elegans, using the pair-wise alignment tool WABA (Kent and Zahler, 2000), revealed that 50.1% of the C. briggsae genome aligns to 52.3% of the C. elegans genome (Stein et al., 2003). Merged WABA alignments, 4,837 in total, cover 84.6% of the C. briggsae genome and 80.8% of the C. elegans genome. Classification of these WABA alignments by searching for different types of breakpoints, which are defined as junctions between adjacent colinear blocks with C. briggsae supercontigs, resulted in 1,384 putative inversion events, 244 putative translocation events, and 2,735 putative transposition events between C. briggsae and C. elegans genomes. By examining C. elegans sequences, matching each end of the breakpoint junctions on C. briggsae supercontigs we found a clear bias toward rearrangements being within the same ancestral chromosome (see Table 2).
Essentially all (96%) of the approximately 1,000 identified trans-spliced operons in C. elegans (Blumenthal and Gleason, 2003) are conserved in C. briggsae (Stein et al., 2003), suggesting that operons are under strong purifying selection pressure. Recent high throughput functional genomic projects including microarray analysis (Kim et al., 2001) and SAGE (serial analysis of gene expression; McKay et al., 2003) have demonstrated that genes within operons in C. elegans have positively correlated gene expression (Chen and Stein, 2006; Lercher et al., 2003). Whether genes within operons in C. briggsae are positively correlated in gene expression remains to be examined.
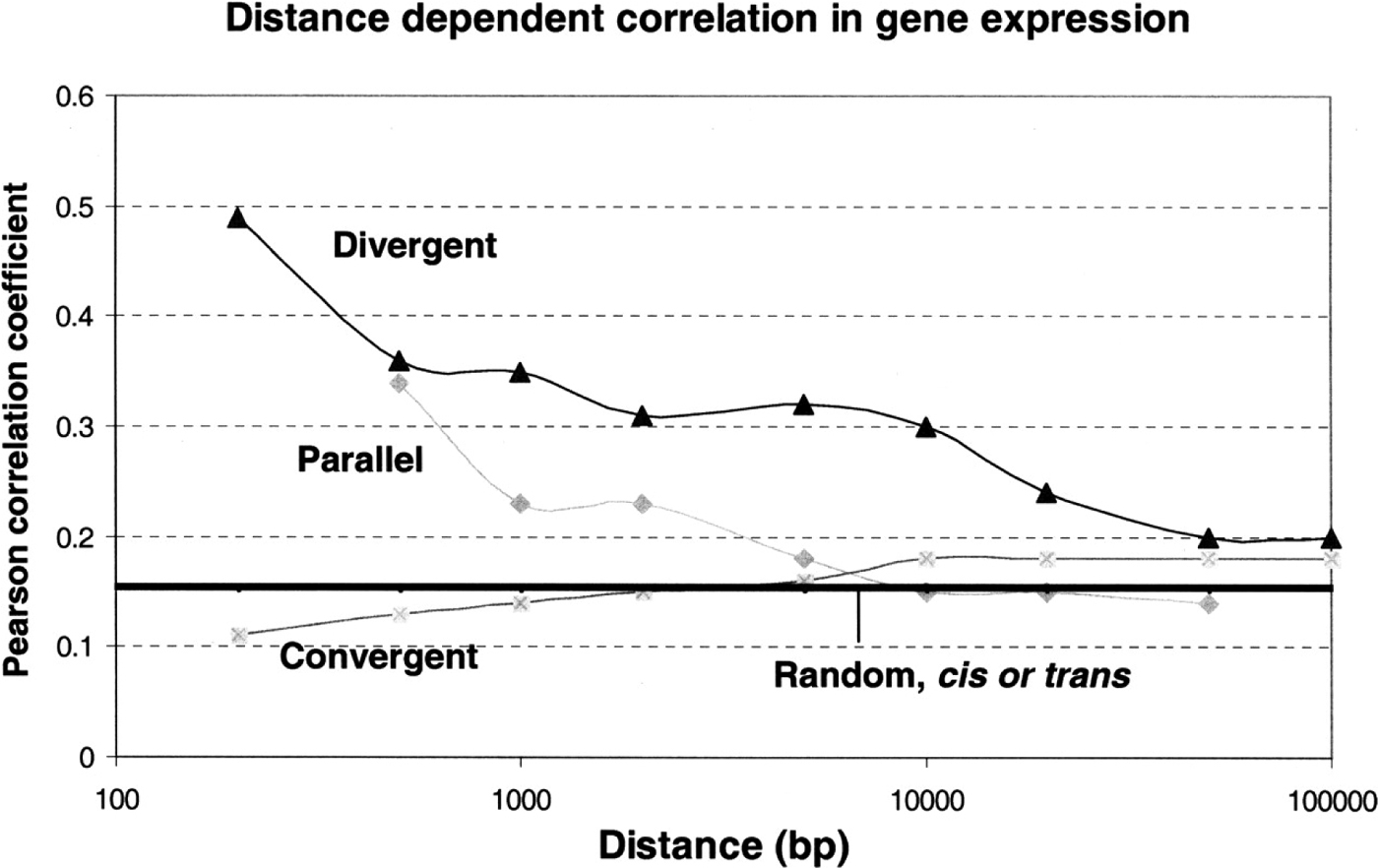
Correlation of gene expression is not limited to genes within operons. Recently, we have demonstrated that expression of neighboring genes is also positively correlated (Chen and Stein, 2006). Using tissue-specific SAGE libraries generated by the C. elegans Gene Expression Consortium, Chen and Stein (Chen and Stein, 2006) found that divergent neighboring gene pairs and parallel neighboring gene pairs are positively correlated in gene expression, while convergent neighboring gene pairs show a greatly reduced correlation in gene expression that is lower than the background correlation (see Figure 2; Chen and Stein, 2006). All of these correlations are dependent on the distances between neighboring gene pairs, the correlations approach background as the distances become longer than 10kbp (see Figure 2). As extreme examples of neighboring genes, we analyzed genes that physically overlap in their gene models (introns, exons and UTRs). Nested gene pairs and convergent gene pairs demonstrated lack of correlation in gene expression, while piggyback gene pairs showed positive correlation in gene expression. Since no large-scale functional genomics projects such as microarray analysis or SAGE have been carried out in C. briggsae, correlation of gene expression for its neighboring genes and overlapping genes are unknown. However, similar types and extends of correlation in gene expression likely exist in C. briggsae because overlapping genes are well conserved between the two nematodes (Chen and Stein, 2006).
There are similar numbers of program-predicted non-coding RNA genes in C. briggsae (962 and 191 fragmentary matches) and C. elegans (838 and 212 fragmentary matches). A detailed list of various types of RNA genes is included in Table 1, which shows that both species have similar number of each type of non-coding RNAs. For example, both species have exactly 18 SL2 spliced leader genes, and 70 out of 105 identified C. elegans miRNAs have close homologs in C. briggsae.
|
Figure 2. Distance-dependent correlation in gene expression. x-axis is the distance between neighboring genes and y-axis is the Pearson correlation coefficient values. (Parallel) Parallel gene pairs; (Divergent) divergent neighboring gene pairs; (Convergent) convergent neighboring gene pairs. Each point in the figure represents a median value for the Pearson correlation coefficient for each group. Pearson correlation coefficient values for cis- and trans-random gene pairs are essentially the same and are represented by a horizontal line.
Most gene families have similar sizes between C. briggsae and C. elegans, as suggested by TRIBE-MCL clustering analysis (Stein et al., 2003). However, there are exceptions in that some gene families are larger in C. briggsae than in C. elegans and vice versa. For example, there are many more putative chemosensory genes in C. elegans than in C. briggsae (Chen et al., 2005; Stein et al., 2003). Breaking down the putative chemosensory superfamily into smaller families, shows that some families have similar sizes in C. briggsae and C. elegans, while others, including the srab family (Chen et al. 2005b) and the srz family (Thomas et al., 2005), have twice as many genes in C. elegans than in C. briggsae. Such gene family size differences resulted largely from massive tandem duplications of individual genes in the C. elegans genome. Tandem duplication were also observed in C. briggsae but both the ratio and frequency of expansion was not as dramatic in C. briggsae as in C. elegans (Chen et al., 2005). Altogether, TRIBE-MCL clustering analysis demonstrated that for gene clusters with five or more C. briggsae and C. elegans genes, there were 202 clusters (corresponding to gene families) with gene number differences of at least 2-fold between C. briggsae and C. elegans (Stein et al., 2003).
Morphologically C. briggsae is almost identical to C. elegans. However, as described above, a comparison of the genome sequences between the two species has revealed a significant number of species-specific genes (~2,000 in total; Stein et al., 2003). Hence, an obvious question is: why are these changes not reflected in significant morphological or behavioral differences? Perhaps such features are under strong selective pressure and have resulted in compensatory changes in the genome. Alternatively, it is possible that the two species exhibit differences in other developmental and physiological processes that have yet to be revealed. In the following subsections we summarize studies focusing on known differences between C. briggsae and C. elegans (as well as other Caenorhabditis nematodes). With increasing research into nematode evolution, future studies are expected to reveal additional differences and the roles of species-specific genes in mediating some of these changes.
The excretory system in C. elegans plays a critical role in osmoregulation. It is made up of just three cells: the canal cell, the duct cell and the pore cell. The duct and pore cells connect the canal to the outside world and help eliminate excess body fluid. In C. briggsae the excretory system looks morphologically identical, however C. briggsae animals have more anterior duct opening than C. elegans (Wang and Chamberlin, 2002). This difference in the placement of duct opening is the result of the altered expression of lin-48, a Drosophila ovo family member (Wang and Chamberlin, 2002). In C. elegans, lin-48 is expressed in the excretory duct cell, a subset of the hindgut cells and a few neurons. In contrast, C. briggsae lin-48 does not express in the duct cell due to an evolutionary change in the regulation of Cbr-lin-48. This demonstrates that evolutionary changes in the regulation of C. briggsae genes (or C. elegans genes) can give rise to morphological differences.
Regulation of sex-specific developmental programs is of prime importance for the propagation and diversity of multicellular organisms. Experimental findings in metazoans have revealed that, in spite of their critical role, many sex determination genes and genetic pathways have rapidly evolved. Because of their morphological diversity and two different modes of reproduction (hermaphroditic, e.g., C. briggsae and C. elegans, and gonochoristic, e.g., C. remanei, C. japonica and C. sp.CB5161), nematodes provide excellent opportunity to study the molecular basis of sexual differentiation and evolution. In this respect, the C. briggsae genome has facilitated identification of the sex-determination genes and study of their functional differences. Given that hermaphroditism has evolved independently in C. elegans and C. briggsae (Kiontke et al., 2004), it is conceivable that the underlying regulatory mechanisms have diverged between the two species. A survey of the C. elegans sex pathway genes in C. briggsae genome has revealed significant sequence divergence in some genes suggesting functional differences in the mode of sex determination. Consistent with this observation, FEM-3 (novel) and TRA-2 (transmembrane domain) proteins have rapidly evolved in C. briggsae and C. remanei (Haag and Ackerman, 2005; Haag et al., 2002). In C. elegans, the two proteins are known to physically interact with each other (Mehra et al., 1999). While this interaction appears to be conserved in C. briggsae, the two proteins function in strictly species-specific manners. Furthermore, RNA interference (RNAi)-mediated inactivation of the fem-1 and fem-2 genes in C. briggsae reveal functional differences from C. elegans (Stothard and Pilgrim, 2003). In a separate study, fog-2, which is required for hermaphrodite spermatogenesis in C. elegans, has been shown to be absent in C. briggsae (Nayak et al., 2005). Hence, the genome sequence of C. briggsae promises new understanding of the evolution of sex-determination pathways.
Nematode vulval development is an established model for comparative developmental biology. In C. elegans, six vulval precursor cells (VPCs) (Pn.p, where n=3–8) respond to a spatially graded inductive signal from the gonadal anchor cell (AC) and give rise to a 3°-3°-2°-1°-2°-3° cell fate pattern. While the 1° and 2° VPCs divide to give rise to vulval tissue, the 3° lineage cells fuse into the surrounding hypodermal syncytial cell (hyp7). The cell ablation experiments have shown that all six VPCs are equally capable of adopting 1° and 2° fates (Horvitz and Sternberg, 1991). For example, ablation of all VPCs but the P3.p, allows it to generate vulval progeny although it does not do so in an intact animal. By contrast, in C. briggsae P3.p does not appear to be competent by this assay (Delattre and Felix, 2001). However, in certain multivulva mutants of C. briggsae P3.p is capable of being induced and give rise to vulval progeny suggesting that negative regulation may restrict the fate of P3.p in wild-type animals (B. Gupta, unpublished). Thus, the competence of P3.p appears to have evolved between the two species.
Rudel and Kimble (Rudel and Kimble, 2001; Rudel and Kimble, 2002) have examined evolution of Notch-family members, lin-12 and glp-1, in Caenorhabditis nematodes (C. elegans, C. briggsae and C. remanei). Their results suggest that in C. briggsae lin-12 and glp-1 have acquired novel functions. Furthermore, there appears to be two lin-12-like genes in C. briggsae compared to only one in C. elegans. In C. elegans LIN-12 and GLP-1 are functionally interchangeable and in wild-type animals perform some overlapping functions, but they are uniquely required in some tissues (see LIN-12/Notch signaling in C. elegans). GLP-1 is necessary during early embryogenesis and later in the development and proliferation of the germline whereas LIN-12, but not GLP-1, is required during lateral signaling events in some somatic tissues such as the vulva. In C. elegans loss of function alleles of lin-12 exhibit protruding vulva (Pvul) phenotype as a result of the fate conversion of the 2° lineage cells P5.p and P7.p into the 1° type cell lineage (see LIN-12/Notch signaling in C. elegans). glp-1 loss of function alleles, on the other hand, do not affect vulval development. However, in C. briggsae, glp-1 is required for vulval development. Cbr-glp-1(RNAi) animals exhibit a multivulva phenotype suggesting that wild-type glp-1 functions to regulate the competence of the VPCs. Likewise examination of the role of lin-12 revealed that, unlike lin-12 in C. elegans, lin-12 in C. briggsae is essential for larval viability. Cbr-lin-12(RNAi) animals arrest during L1 larval stage, a phenotype that is not observed in Cel-lin-12(RNAi) animals. Functional differences between lin-12 and glp-1 in C. elegans and C. briggsae are supported by the low level of amino acid conservation (68% and 61% identities, respectively) and may result from evolutionary changes in protein-protein interactions.
In a separate study, Kirouac and Sternberg (Kirouac and Sternberg, 2003) have examined regulation of the three orthologs that are expressed in vulval cells (egl-17, zmp-1 and cdh-3) in C. briggsae and have identified many key regulatory elements that are conserved between the two species. In spite of this conservation, the dynamics of the expression of these genes in vulval cells have evolved. Hence, for example, the C. elegans egl-17::GFP reporter, which is always expressed in vulC and vulD cell types in C. elegans, often fails to express in the same set of cells in C. briggsae. The expression pattern of the cdh-3::GFP reporter, on the other hand, is significantly diverged in C. briggsae (Kirouac and Sternberg, 2003). This finding is somewhat similar to the regulatory differences reported for the pes-1 and fkh-2 forkhead family members (Molin et al., 2000). While both these genes are functionally redundant in C. elegans and C. briggsae embryogenesis, C. briggsae fkh-2 has acquired additional novel function.
The male tail, involved in copulation, exhibits considerable differences in morphology among the Rhabditidae family (including C. elegans and C. briggsae). This variability is one criteria used by researchers to distinguish between and classify various nematodes (Fitch, 1997). C. briggsae's male tail has a subtle difference, relative to C. elegans, in its pattern of ray morphology, specifically there is a tendency of ray 3 to fuse with ray 4. Baird and colleagues (Baird et al., 2005) have used recombinant inbred lines (RILs) to map at least two loci (linked to Cbr-unc-22) that appear to play a role in this morphological variation.
Studies focusing on the ability of nematodes to survive in different environmental conditions have shown evolutionary differences between C. elegans and C. briggsae. Examination of the thermal sensitivity of C. elegans has revealed significant reduction in the brood size at temperatures above 25°C and almost complete sterility above 27°C (M. Ailion and J. Thomas, personal communication; B. Gupta, unpublished). By contrast, C. briggsae grows well at 27.5°C with no significant reduction in brood size (AF16: 200 +/− 35 compared to 234 +/− 25 at 20°C; Fodor et al., 1983). In fact, C. briggsae can grow and develop to adulthood at 30°C (M. Ailion and J. Thomas, personal communication; RJ, unpublished). These studies have been done using multiple isolates of C. elegans and C. briggsae and all isolates of each species behave similarly. The formation of dauer larva, a developmentally arrested diapause stage, is another temperature-dependent trait that exhibits differences between C. briggsae and other Caenorhabditis nematodes (M. Ailion and J. Thomas, personal communication; T. Inoue and P. Sternberg, personal communication). C. elegans when grown at temperature above 25°C forms dauers even when well-fed, and analysis of mutants show these temperatures to be a strong dauer-inducing condition. In contrast, well-fed C. briggsae does not form dauers at these temperatures and analysis of mutants have failed to detect temperature-induced dauer induction at temperatures above 25°C (T. Inoue, personal communication).
In multicellular organisms many cells and tissues exhibit morphological and functional asymmetries. C. elegans has been successfully used to study function of some of the genes that confer asymmetry on neural cells (Hobert et al., 2002). Ortiz and colleagues (Ortiz et al., 2006) have carried out a detailed study of the sequence and expression pattern of the guanylyl cyclase (gcy) genes in C. elegans and C. briggsae. A total of 23 (out of 33) C. briggsae gcy genes have clear one-to-one orthologs in C. elegans whereas the non-orthologous family members (10 in C. briggsae and 11 in C. elegans) appear to have arisen as a result of species-specific gene duplication events. The expression patterns of two gcy genes (gcy-4 and gcy-19) in C. briggsae ASE neurons (ASEL and ASER) show striking evolutionary differences that resulted from changes in cis-acting elements and trans-acting factors. Since the C. elegans ASE neurons exhibit functional left/right asymmetry and express distinct set of gcy genes, the mechanism controlling regulation of the ASE laterality appears to have evolved in C. briggsae.
The developmental and behavioral differences between C. briggsae and C. elegans summarized above are just the beginning. With increasing number of laboratories focusing on briggsae-elegans comparative studies, many more differences are likely to be revealed in future. This argues for a need to develop resources and tools to facilitate experimental studies in C. briggsae.
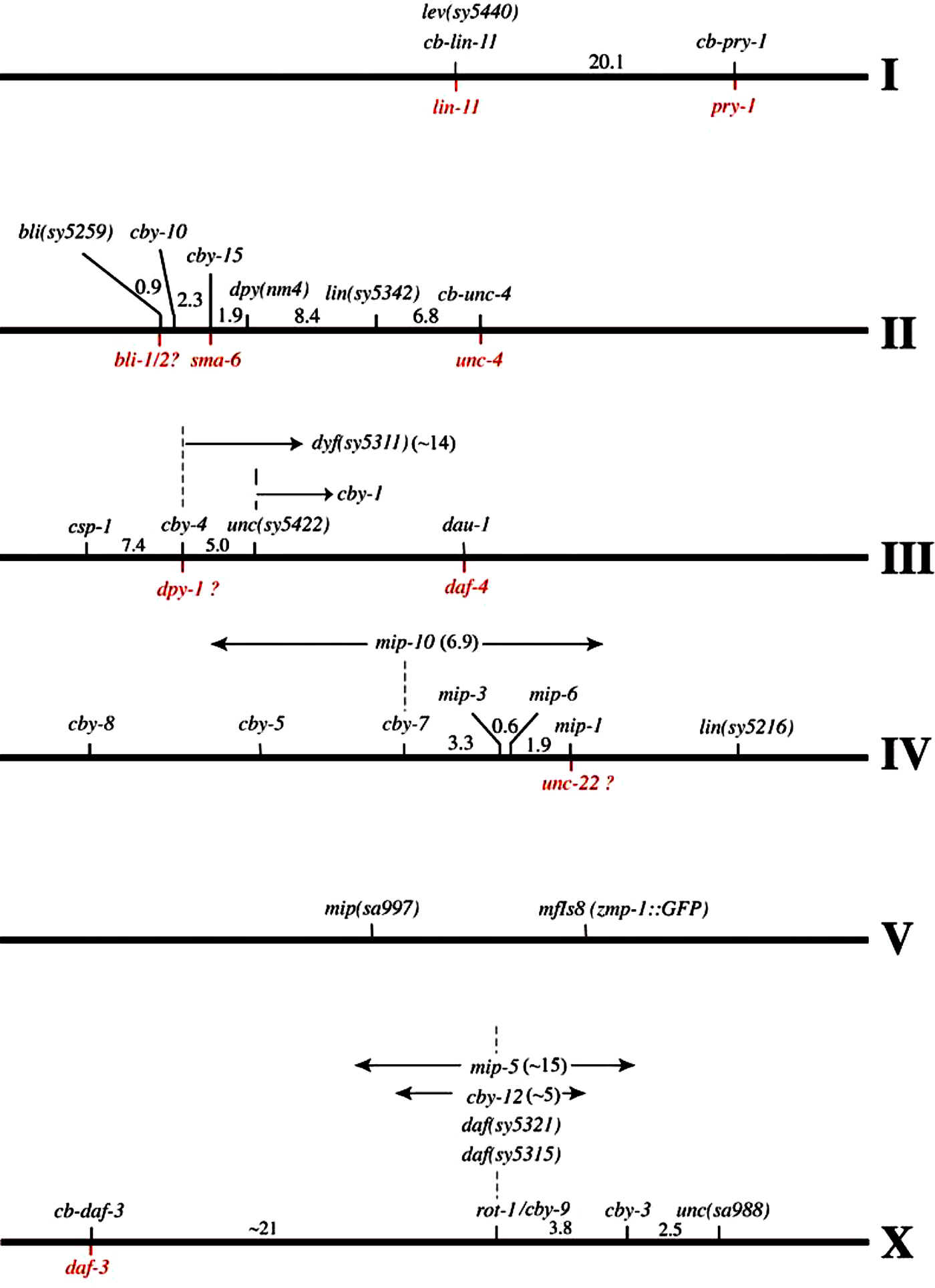
Given the availability of the C. briggsae genome sequence, RNAi could be one approach to examine the role of known genes in a given biological process. The success of the RNAi approach in C. elegans has made it a method of choice for large-scale functional studies in other nematodes. However, it should be pointed out that while RNAi-based screen is likely to identify some of the functional genes, factors such as weak RNAi penetrance, pleotropy and genetic redundancy might not reveal function of many genes. If there is a difference in the RNAi phenotype of a gene it is unclear if it is a quantitative difference in the use of that gene, or in its susceptibility to RNAi. In this respect, genetic approaches promise success. Studies of genetic mutations have revealed a spectrum of phenotypes associated with hypomorphic (loss-of-function), hypermorphic (gain-of-function) and neomorphic (novel) alleles. Achieving these types of results are not possible by using standard RNAi or gene knock-out screens. Highly evolving genes (e.g., GPCR family, sex-determination genes) may have acquired novel functions that can best be identified by isolating genetic mutations, then studying their phenotypes and genetic interactions. To facilitate forward genetic experiments in C. briggsae, mutations with visible phenotypes such as Unc (Uncoordinated), Dpy (Dumpy) and Rol (Roller) have been isolated in different laboratories (B. Gupta, P. Sternberg and D. Baillie, unpublished; Table 3; the C. briggsae website). A number of these have been mapped leading to the definition of six independent linkage groups (see Figure 3; the C. briggsae website). The linkage map will facilitate genetic analysis of the C. briggsae genes. Some of the studies that are currently ongoing involve development of the vulva (B. Gupta, unpublished), sex determination (Hill et al., 2006), ray pattern formation (Baird et al., 2005), and the excretory system features and salt tolerance behavior (H. Chamberlin, personal communication).
Table 3. The list of genetically characterized mutations in C. briggsae
| Locus | AKA | LG | Reference allele | Alleles | Phenotype |
|---|---|---|---|---|---|
| bli(sy5259) | II | sy5259 | 1 | Blister along the entire body, symmetrical | |
| Cbr-daf-3 | X | sy5417 | 1 | daf-d | |
| Cbr-daf-4 | dau-1 | III | sa973 | 1 | ts for small size and dauer (daf-c) at 27°C |
| dpy(s1281) | cby-1 | III | s1281 | 3 | Strong Dpy |
| dpy(s1282) | cby-2 | X | s1282 | 1 | Strong Dpy, slightly paralyzed |
| dpy(sy5039) | cby-3 | X | sy5039 | 1 | Strong Dpy |
| dpy(s1272) | cby-4 | III | s1272 | 19 | Strong Dpy |
| dpy(s1286) | cby-5 | IV | s1286 | 1 | Medium Dpy |
| dpy(sy5027) | cby-7 | IV | sy5027 | 2 | Medium Dpy, weak roller |
| dpy(sy5017) | cby-8 | IV | sy5017 | 4 | Medium Dpy, slow moving |
| dpy(sy5048) | cby-9; rot-1 | X | sy5048 | 2 | Medium Dpy, Adults are weak rollers |
| dpy(sy5064) | cby-10 | II | sy5064 | 2 | Strong Dpy, very slow and often sick |
| dpy(sy5057) | cby-12 | X | sy5057 | 1 | Medium Dpy |
| dpy(sy5005) | cby-13 | sy5005 | 3 | Strong Dpy, weakly Egl, weakly Rol when backing | |
| dpy(sy5076) | cby-14 | sy5076 | 5 | Medium Dpy | |
| dpy(sy5025) | cby-16 | sy5025 | 1 | Strong Dpy | |
| dpy(sy5075) | cby-17 | sy5075 | 2 | Strong Dpy, poor moving | |
| dpy(sa971) | X | sa971 | 1 | Strong Dpy | |
| dpy(sa995) | X | sa995 | 1 | Medium Dpy | |
| dpy(sa996) | IV | sa996 | 1 | Medium Dpy | |
| dpy(nm3) | X | nm3 | 1 | Strong Dpy | |
| dpy(nm4) | II | nm4 | 1 | Strong Dpy | |
| lev(sy5436) | I | sy5436 | 1 | Resistant to 1mM levamisole, Unc | |
| lev(sy5437) | I | sy5437 | 3 | Resistant to 1mM levamisole, Unc | |
| lev(sy5440) | I | sy5440 | 1 | Resistant to 1mM levamisole, Unc | |
| Cbr-lin-11 | I | sy5336 | 2 | Pvul, Egl | |
| lin(sy5216) | IV | sy5216 | 2 | Multivulva | |
| lin(sy5342) | II | sy5342 | 2 | Multivulva | |
| lon(sy5227) | X | sy5227 | 1 | Thin and long, males mate poorly | |
| Cbr-pry-1 | I | sy5353 | 2 | Multivulva | |
| rol(sy5143) | rot-2 | X | sy5143 | 1 | Right-handed roller |
| rol(sy5412) | sy5412 | 1 | All stages right-handed roller | ||
| Cbr-sma-6 | cby-15 | II | sy5148 | 1 | Strong Dpy, Small size, fairly active |
| sma(sa985) | sa985 | 1 | Small size, active | ||
| sma(sy5002) | sy5002 | 2 | Small size, active | ||
| sma(sy5159) | X | sy5159 | Small size | ||
| unc(s1270) | mip-1 | IV | s1270 | 11 | Twicher, thin |
| unc(ge19) | mip-3 | IV | ge19 | 1 | Almost paralyzed, tends to coil |
| unc(sy5077) | mip-5 | X | sy5077 | 1 | almost paralyzed, kinker |
| unc(sy5019) | mip-6 | IV | sy5019 | 1 | A coiler, weak kinker, paralyzed |
| unc(sa984) | mip-10 | IV | sa984 | 1 | Slow moving, weak Dpy |
| Cbr-unc-4 | II | sy5341 | 1 | Active, fails to back when touched | |
| unc(sa972) | csp-1 | III | sa972 | 1 | Slow moving, defective in backward movement, slightly smaller than wild-type |
| unc(sa986) | V | sa986 | 1 | kinker, slight curler | |
| unc(sa987) | IV | sa987 | 1 | Medium Dpy | |
| unc(sa988) | X | sa988 | 1 | Kinker, weak Dpy | |
| unc(sa997) | V | sa997 | 1 | Slightly thin, slow movement in adults | |
| unc(sy5094) | V | sy5094 | 1 | Kinker, curler | |
| unc(sy5329) | X | sy5329 | 1 | Medium Unc, poor in backward movement | |
| unc(sy5422) | III | sy5422 | 1 | Strong Unc, small size, does not form dauer |
Several tools are currently available to study evolutionary changes in the function of C. briggsae genes and carry out C. briggsae-C. elegans comparative studies.
Among different approaches to deliver double-stranded RNA (dsRNA) to intact C. elegans cells, RNAi by feeding (systemic RNAi) is a rapid method to study gene function (Timmons et al., 2001). While the RNAi by microinjection technique has been successfully used to examine function of some of the genes in C. briggsae (Stothard and Pilgrim, 2003), wild-type C. briggsae AF16 is resistant to systemic RNAi. This is due to the lack of a functional sid-2 gene since the AF16 worms expressing C. elegans sid-2 (Cel-sid-2) are susceptible to systemic RNAi (C. Hunter, personal communication). Hence, a C. briggsae transgenic strain carrying integrated Cel-sid-2 array can be used in functional genomic studies.
|
Figure 3. The phenotypic marker-based genetic linkage map of C. briggsae (v9.0). The C. elegans orthologs, that have been experimentally confirmed, are shown below the lines. The question marked loci represent candidates based on the genetic properties (phenotype, allele frequency and map position) and require further confirmation.
To facilitate genetic studies of C. briggsae, a large collection of mutant strains (more than 200) is freely available to the community (see Table 3). These strains not only allow genetic and molecular analysis of the involved loci but also serve as useful markers in phenotypic analyses and linkage mapping of new mutants. While most of these are curated in three different laboratories (B. Gupta, P. Sternberg and D. Baillie), some of the strains are available at the Caenorhabditis Genetics Center for centralized distribution. A comprehensive list of the mutants can be found at the C. briggsae website.
The phenotypic marker-based genetic linkage map is currently under construction (see Figure 3; also see the C. briggsae website; B. Gupta, T. Inoue, RJ, A. Mah, D. Ballie and P. Sternberg, unpublished). This has involved standard 2 and 3-factor genetic mapping of mutations that exhibit visible phenotypes and cloning of some of the loci. In parallel, R. Waterston and colleagues (personal communication) are working to construct a single nucleotide polymorphism (SNP)-based genetic linkage map of C. briggsae. This approach involves careful mapping of SNPs identified by partial sequencing of two C. briggsae isolates HK104 and VT847. The current version of the SNP-based genetic linkage map (v3.1) is based on the 248 genotyped SNPs that are roughly equally distributed over the six chromosomes. Integration of the SNP-based and mutant-based maps, which is ongoing (B. Gupta and R. Miller, unpublished), will allow researchers to place their gene of interest on different linkage groups, examine synteny with C. elegans, carry out epistasis and genetic interaction experiments. This will facilitate cloning of the C. briggsae genes, in particular those with the novel mutant phenotypes i.e., ones for which there are no candidate genes proposed from our knowledge of C. elegans, and study evolutionary changes in gene function.
Mos1 is a Drosophila transposon that has been successfully used in C. elegans to generate mutations (Bessereau et al., 2001). The advantage of this system is that it can be used as a sequence tag to rapidly identify the mutated gene by PCR and therefore circumvent the need for time-consuming genetic mapping and gene cloning experiments. Dr. Marie-Anne Felix has tested the applicability of the system in C. briggsae and successfully recovered mutations in three genes (http://snp.wustl.edu/snp-research/c-briggsae/mos-mutagenesis.html). While the experimental protocol is in the early phase of development in C. briggsae, it has potential of rapidly isolating alleles of the C. elegans orthologs (M.-A. Felix, personal communication; B. Gupta, unpublished).
The wild populations of C. briggsae and C. elegans serve as useful resources for studies involving natural variation and ecology of the species. Comparison of the genomic DNA sequences in these strains has shown greater nucleotide diversity in C. briggsae compared to C. elegans (Graustein et al., 2002; Jovelin et al., 2003). Currently more than 60 wild isolates of C. briggsae are available, they have been collected from various regions of the world (S. Baird and M.-A. Felix, personal communications). These include 57 new isolates that are part of a recent study looking at the pattern of polymorphisms in 63 strains across the three continents (Asia, Europe and America; Cutter et al., 2006). In addition, a large number of recombinant inbred lines (RILs) have been generated. These are derived from two sets of crosses between AF16, HK104 and VT847 (set 1: AF16 and HK104, set 2: AF16 and VT847; S. Baird, personal communication). Besides their use in mapping quantitative trait loci (QTLs), the RILs are playing key role in construction of the SNP-based genetic linkage map (R. Miller and S. Baird, personal communications).
The genomic clones of C. briggsae (in the form of BAC and fosmid clones) were generated to construct a high resolution physical map to help assemble the sequenced genome. The BAC clones are available from the Children's Hospital Oakland Research Institute.
We are grateful to David Baillie, Marie-Anne Felix, Craig Hunter, Raymond Miller, Scott Baird, Helen Chamberlin, Michael Ailion, James Thomas, Takao Inoue and Robert Waterston for sharing their unpublished work. We thank David Baillie for sharing his ideas and reviewing the manuscript. B. Gupta is a tier II Canada Research Chair and supported by grants from NSERC and NIH. NC is supported by a grant from NSERC, a faculty startup fund from Simon Fraser University, and the Simon Fraser University President's Research Grants Fund. RJ is supported by NSERC and CIHR.
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. Abstract Article
Baird, S.E., Davidson, C.R., and Bohrer, J.C. (2005). The genetics of ray pattern variation in Caenorhabditis briggsae. BMC Evol. Biol. 5, 3. Abstract Article
Bessereau, J.L., Wright, A., Williams, D.C., Schuske, K., Davis, M.W., and Jorgensen, E.M. (2001). Mobilization of a Drosophila transposon in the Caenorhabditis elegans germ line. Nature 413, 70–74. Abstract Article
Blumenthal, T., and Gleason, K.S. (2003). Caenorhabditis elegans operons: form and function. Nat. Rev. Genet. 4, 112–120. Abstract Article
Chen, N., Harris, T.W., Antoshechkin, I., Bastiani, C., Bieri, T., Blasiar, D., Bradnam, K., Canaran, P., Chan, J., Chen, C.K., et al. (2005). WormBase: a comprehensive data resource for Caenorhabditis biology and genomics. Nucleic Acids Res. 33, D383–389. Abstract Article
Chen, N., Pai, S., Zhao, Z., Mah, A., Newbury, R., Johnsen, R.C., Altun, Z., Moerman, D.G., Baillie, D.L., and Stein, L.D. (2005). Identification of a nematode chemosensory gene family. Proc. Natl. Acad. Sci. U.S.A. 102, 146–151. Abstract Article
Chen, N., and Stein, L.D. (2006). Conservation and functional significance of gene topology in the genome of Caenorhabditis elegans. Genome Res. 16, 606–617. Abstract Article
Cho, S., Jin, S.W., Cohen, A., and Ellis, R.E. (2004). A phylogeny of caenorhabditis reveals frequent loss of introns during nematode evolution. Genome Res. 14, 1207–1220. Abstract Article
Coghlan, A., and Wolfe, K.H. (2002). Fourfold faster rate of genome rearrangement in nematodes than in Drosophila. Genome Res. 12, 857–867. Abstract Article
Coghlan, A., and Wolfe, K.H. (2004). Origins of recently gained introns in Caenorhabditis. Proc. Natl. Acad. Sci. U.S.A. 101, 11362–11367. Abstract Article
Cutter, A.D., Félix, M.A., Barrière, A., and Charlesworth, D. (2006). Patterns of nucleotide polymorphism distinguish temperate and tropical wild isolates of Caenorhabditis briggsae. Genetics in press.
Delattre, M., and Felix, M.A. (2001). Polymorphism and evolution of vulval precursor cell lineages within two nematode genera, Caenorhabditis and Oscheius. Curr. Biol. 11, 631–643. Abstract Article
Fitch, D.H. A. (1997). Evolution of male tail morphology and development in rhabditid nematodes related to Caenorhabditis elegans. Syst. Biol. 46, 145–179. Abstract Article
Fodor, A., Riddle, D.L., Nelson, F.K., and Golden, J.W. (1983). Comparison of a new wild-type Caenorhabditis briggsae with laboratory strains of C. briggsae and C. elegans. Nematologica 29, 203–217.
Graustein, A., Gaspar, J.M., Walters, J.R., and Palopoli, M.F. (2002). Levels of DNA polymorphism vary with mating system in the nematode genus caenorhabditis. Genetics 161, 99–107. Abstract
Haag, E.S., and Ackerman, A.D. (2005). Intraspecific variation in fem-3 and tra-2, two rapidly coevolving nematode sex-determining genes. Gene 349, 35–42. Abstract Article
Haag, E.S., Wang, S., and Kimble, J. (2002). Rapid coevolution of the nematode sex-determining genes fem-3 and tra-2. Curr. Biol. 12, 2035–2041. Abstract Article
Heschl, M.F., and Baillie, D.L. (1990). Functional elements and domains inferred from sequence comparisons of a heat shock gene in two nematodes. J. Mol. Evol. 31, 3–9. Abstract Article
Hill, R.C., de Carvalho, C.E., Salogiannis, J., Schlager, B., Pilgrim, D., and Haag, E.S. (2006). Genetic flexibility in the convergent evolution of hermaphroditism in Caenorhabditis nematodes. Dev. Cell 10, 531–538. Abstract Article
Hillier, L.W., Coulson, A., Murray, J.I., Bao, Z., Sulston, J.E., and Waterston, R.H. (2005). Genomics in C. elegans: so many genes, such a little worm. Genome Res. 15, 1651–1660. Abstract Article
Hobert, O., Johnston, R.J., Jr., and Chang, S. (2002). Left-right asymmetry in the nervous system: the Caenorhabditis elegans model. Nat. Rev. Neurosci. 3, 629–640. Abstract Article
Horvitz, H.R., and Sternberg, P.W. (1991). Multiple intercellular signalling systems control the development of the Caenorhabditis elegans vulva. Nature 351, 535–541. Abstract Article
Jovelin, R., Ajie, B.C., and Phillips, P.C. (2003). Molecular evolution and quantitative variation for chemosensory behaviour in the nematode genus Caenorhabditis. Mol. Ecol. 12, 1325–1337. Abstract Article
Kennedy, B.P., Aamodt, E.J., Allen, F.L., Chung, M.A., Heschl, M.F., and McGhee, J.D. (1993). The gut esterase gene (ges-1) from the nematodes Caenorhabditis elegans and Caenorhabditis briggsae. J. Mol. Biol. 229, 890–908. Abstract Article
Kent, W.J., and Zahler, A.M. (2000). Conservation, regulation, synteny, and introns in a large-scale C. briggsae-C. elegans genomic alignment. Genome Res. 10, 1115–1125. Abstract Article
Kim, S.K., Lund, J., Kiraly, M., Duke, K., Jiang, M., Stuart, J.M., Eizinger, A., Wylie, B.N., and Davidson, G.S. (2001). A gene expression map for Caenorhabditis elegans. Science 293, 2087–2092. Abstract Article
Kiontke, K., Gavin, N.P., Raynes, Y., Roehrig, C., Piano, F., and Fitch, D.H. (2004). Caenorhabditis phylogeny predicts convergence of hermaphroditism and extensive intron loss. Proc. Natl. Acad. Sci. U.S.A. 101, 9003–9008. Abstract Article
Kirouac, M., and Sternberg, P.W. (2003). cis-Regulatory control of three cell fate-specific genes in vulval organogenesis of Caenorhabditis elegans and C. briggsae. Dev. Biol. 257, 85–103. Abstract Article
Lee, Y.H., Huang, X.Y., Hirsh, D., Fox, G.E., and Hecht, R.M. (1992). Conservation of gene organization and trans-splicing in the glyceraldehyde-3-phosphate dehydrogenase-encoding genes of Caenorhabditis briggsae. Gene 121, 227–235. Abstract Article
Lercher, M.J., Blumenthal, T., and Hurst, L.D. (2003). Coexpression of neighboring genes in Caenorhabditis elegans is mostly due to operons and duplicate genes. Genome Res. 13, 238–243. Abstract Article
Marra, M.A., Kucaba, T.A., Dietrich, N.L., Green, E.D., Brownstein, B., Wilson, R.K., McDonald, K.M., Hillier, L.W., McPherson, J.D., and Waterston, R.H. (1997). High throughput fingerprint analysis of large-insert clones. Genome Res. 7, 1072–1084. Abstract
McKay, S.J., Johnsen, R., Khattra, J., Asano, J., Baillie, D.L., Chan, S., Dube, N., Fang, L., Goszczynski, B., Ha, E., et al. (2003). Gene expression profiling of cells, tissues, and developmental stages of the nematode C. elegans. Cold Spring Harb. Symp. Quant. Biol. 68, 159–169. Abstract Article
Mehra, A., Gaudet, J., Heck, L., Kuwabara, P.E., and Spence, A.M. (1999). Negative regulation of male development in Caenorhabditis elegans by a protein-protein interaction between TRA-2A and FEM-3. Genes Dev. 13, 1453–1463. Abstract
Molin, L., Mounsey, A., Aslam, S., Bauer, P., Young, J., James, M., Sharma-Oates, A., and Hope, I.A. (2000). Evolutionary conservation of redundancy between a diverged pair of forkhead transcription factor homologues. Development 127, 4825–4835. Abstract
Nayak, S., Goree, J., and Schedl, T. (2005). fog-2 and the evolution of self-fertile hermaphroditism in Caenorhabditis. PLoS Biol. 3, e6. Abstract Article
O'Brien, K.P., Remm, M., and Sonnhammer, E.L. (2005). Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 33, D476–480. Abstract Article
Ortiz, C.O., Etchberger, J.F., Posy, S.L., Frokjaer-Jensen, C., Lockery, S., Honig, B., and Hobert, O. (2006). Searching for neuronal left/right asymmetry: genomewide analysis of nematode receptor-type guanylyl cyclases. Genetics 173, 131–149. Abstract Article
Prasad, S.S., and Baillie, D.L. (1989). Evolutionarily conserved coding sequences in the dpy-20-unc-22 region of Caenorhabditis elegans. Genomics 5, 185–198. Abstract Article
Robertson, H.M. (1998). Two large families of chemoreceptor genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae reveal extensive gene duplication, diversification, movement, and intron loss. Genome Res. 8, 449–463. Abstract
Robertson, H.M. (2000). The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 10, 192–203. Abstract Article
Robertson, H.M. (2001). Updating the str and srj (stl) families of chemoreceptors in Caenorhabditis nematodes reveals frequent gene movement within and between chromosomes. Chem. Senses 26, 151–159. Abstract Article
Rudel, D., and Kimble, J. (2001). Conservation of glp-1 regulation and function in nematodes. Genetics 157, 639–654. Abstract
Rudel, D., and Kimble, J. (2002). Evolution of discrete Notch-like receptors from a distant gene duplication in Caenorhabditis. Evol. Dev. 4, 319–333. Abstract Article
Stein, L.D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M.R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al. (2003). The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1, E45. Abstract Article
Stothard, P., and Pilgrim, D. (2003). Sex determination gene and pathway evolution in nematodes. Bioessays 25, 221–231. Abstract Article
Thomas, J.H., Kelley, J.L., Robertson, H.M., Ly, K., and Swanson, W.J. (2005). Adaptive evolution in the SRZ chemoreceptor families of Caenorhabditis elegans and Caenorhabditis briggsae. Proc. Natl. Acad. Sci. U.S.A. 102, 4476–4481. Abstract Article
*Edited by Jonathan Hodgkin. Last revised January 10, 2007. Published May 3, 2007. This chapter should be cited as: Gupta, B.P., et al. Genomics and biology of the nematode Caenorhabditis briggsae (May 3, 2007), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.136.1, http://www.wormbook.org.
Copyright: © 2007 Bhagwati P. Gupta, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: guptab@mcmaster.ca
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.