Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Physiological methods entered the world of C. elegans, a model system used for many years to study development and a plethora of biological processes mainly employing genetic, molecular and anatomical techniques. One of the methods introduced by physiologists is the use of Xenopus oocytes for expression of C. elegans ion channels. Oocytes of the South African frog Xenopus laevis are used widely for the expression of mammalian channels and transporters contributing to numerous discoveries in these fields. They now promise to aid C. elegans researchers in deciphering mechanisms of channels function and regulation with implications for mammalian patho-physiology. Heterologous cRNA can be easily injected into Xenopus oocytes and translated proteins can be studied using several techniques including electrophysiology, immunocytochemistry and protein biochemistry. This chapter will focus on techniques used for oocyte preparation and injection, and will give a brief overview of specific methods. Limitations of the use of Xenopus oocytes will be also discussed.
In 1971 Gurdon and colleagues demonstrated that oocytes extracted from the South-African clawed frog Xenopus laevis were able to synthesize hemoglobin consequent to injection of the corresponding mRNA (Gurdon et al., 1971). Indeed oocytes are particularly well suited for expression of proteins following mRNA injection, since they contain accumulated stores of enzymes, organelles and proteins that are normally used after fertilization that can be recruited for heterologous proteins (Gurdon, 1973; Gurdon et al., 1971; Gurdon et al., 1973).
Beside their ability to efficiently translate exogenous mRNA into proteins, Xenopus oocytes have other experimental advantages: 1) X. laevis is easily maintained in captivity, making it a low-cost laboratory animal, 2) oocytes can be readily harvested and, because of their large diameter (1-1.2 mm), are straightforward to injected, 3) frog oocytes are built to survive for long periods of time outside the body and need little more than fresh water, 4) oocytes express a low number of endogenous membrane transporters and channels because they are virtually independent from exogenous nutrients, 5) multiple species of mRNA can be simultaneously injected to study protein complexes formed by multiple subunits, an advantage over transfected mammalian cells for studying large multi-protein complexes, and 6) standard electrophysiological techniques are easily applicable.
Early studies using Xenopus oocytes dealt with proteins that are of little interest to electrophysiologists such as globin, interferon and viral proteins (Gurdon et al., 1974; Laskey and Gurdon, 1973; Laskey and Gurdon, 1974; Laskey et al., 1972; Woodland et al., 1974). In 1982, Miledi and colleagues first demonstrated that Xenopus oocytes could express various ion channels (Miledi et al., 1982a; Miledi et al., 1982b), and oocytes quickly became a preferred method for electrophysiological analysis of channels, receptors and transporters. Electrophysiological studies that employ Xenopus oocytes include: 1) analysis of channel permeability properties and pharmacological sensitivity, 2) investigation of the properties of mutant channel subunits to address structure-function issues, 3) analysis of channel post-translational modifications, assembly and stoichiometry, 4) study of channel modulation by receptors and second messengers, 5) functional screens for genes encoding ion channels and transporters (expression-cloning).
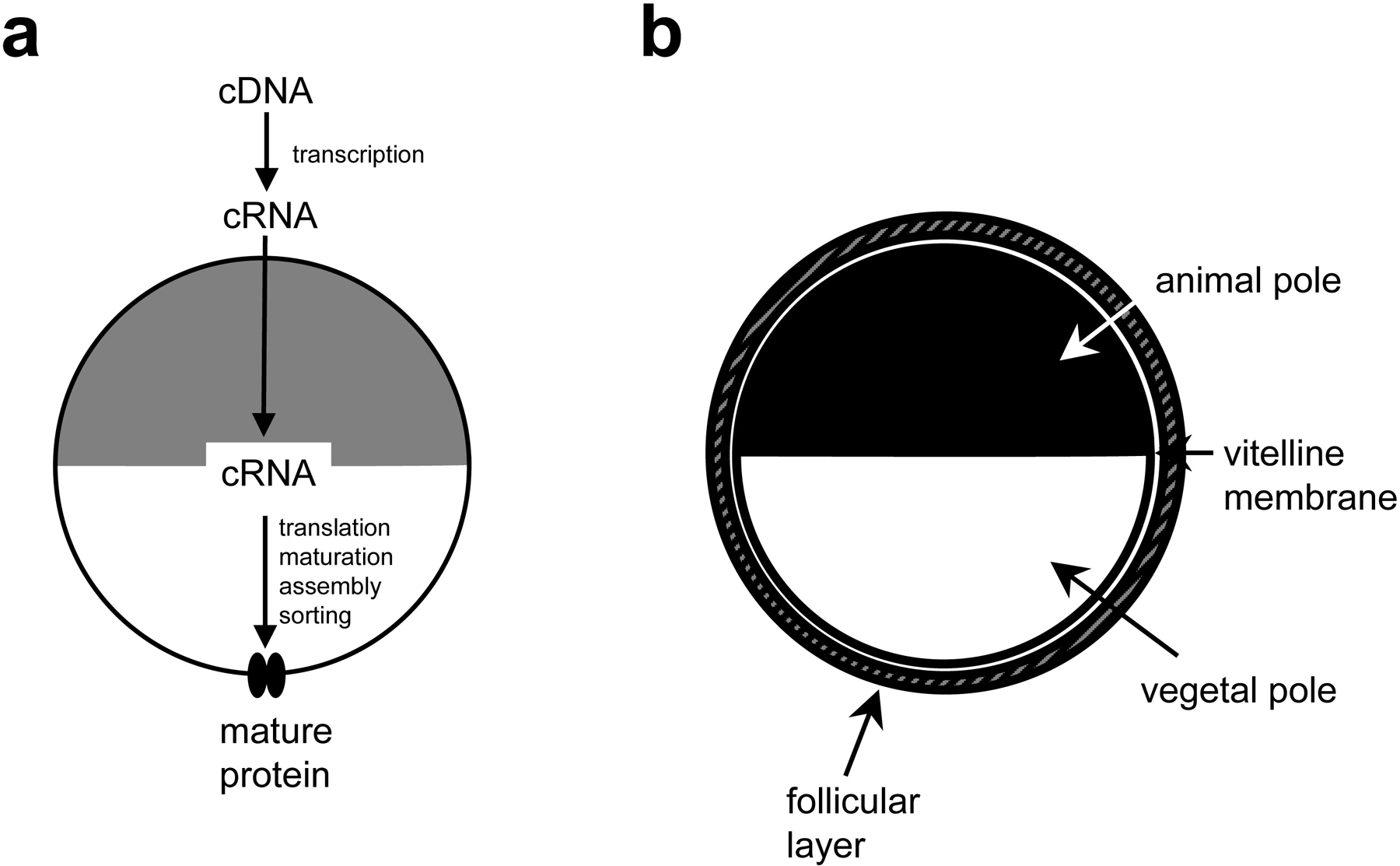
The most common approach for expression of exogenous proteins in Xenopus oocytes is to inject in vitro transcribed complementary RNA into the oocyte cytoplasm (Figure 1a). Expression can be also achieved by injection of the cDNA into the oocyte nucleus (Wickens et al., 1980), but this method requires visual localization of the nucleus and involves the risk of damaging the nuclear membrane during injection; which renders this approach more technically challenging and less efficient. The method that will be reviewed in this chapter is the one based on microinjection of complementary RNA (cRNA) into the oocyte cytoplasm (Figure 1a). Protocols that follow are personally used by the authors. Modifications specific to experimental considerations are discussed in numerous excellent reviews that are recommended for further reading (Tapper and George, 2003; Shih et al., 1998; Stefani, 1998; Stuhmer, 1998; Theodoulou and Miller, 1995; Wang et al., 1991).
|
Figure 1. (a) Cartoon representing the process of expressing exogenous proteins in Xenopus oocytes. (b) A stage V or VI oocyte as found in an ovarian lobe and as it looks following collagenase treatment (2 mg/ml, 1+1 hour at room temperature). When ovarian lobes are treated with 2.5 mg/ml collagenase for 2 hours, they release oocytes devoid of the follicular layer.
A Xenopus oocyte is an immature egg that under the appropriate hormonal stimulation can become competent for fertilization. Oocytes are stored in the abdominal cavity of adult females in clumps called ovarian lobes that include oocytes, connective tissue, blood vessels and follicle cells. Oocytes can be removed by surgical procedures up to three times from the same female. Oocytes go through six maturation stages (I-VI); stage V and VI oocytes are generally used for electrophysiological studies and are the largest (~1.2 mm in diameter). Stage IV oocytes are sometimes preferred for studies of ionic currents with very fast kinetics, because they are smaller and allow a much better temporal clamp.
Oocytes are large cells (1-1.2 mm diameter) with a black pigmented region called the animal pole and a white (non-pigmented) vegetal pole. The oocyte plasma membrane is surrounded by a vitelline membrane, that helps to maintain the oocyte shape and renders the oocyte more resistant to manipulations. The vitelline membrane does not affect electrophysiological recordings since it is devoid of channels and transporters and has a large-enough mesh to allow permeation of ions and small molecules. However, for single channel recordings the vitelline membrane is removed because it hinders the formation of high resistance seal between the patch pipette and the oocyte membrane. Around the vitelline membrane is a layer of follicular cells that separates the oocyte from the external environment (Figure 1b). By contrast to the vitelline membrane, the follicular cells express ion channels and transporters (Miledi and Woodward, 1989a; Miledi and Woodward, 1989b), they are electrically coupled to each other and to the oocyte by gap junctions (Browne and Werner, 1984) and therefore can create serious interference during electrophysiological recordings. For these reasons this layer of cells is eliminated prior to injection by a combination of collagenase treatment and manual stripping.
WormBase (www.wormbase.org) reports the predicted or experimentally-confirmed cDNA sequence for all C. elegans genes. When a cDNA sequence has not been experimentally confirmed though, it is important to keep in mind that less conserved regions (usually 5′ and 3′ end) may be mispredicted. Moreover, splice variants with different functional properties or cell-specific expression may exist and should be considered whenever attempting to fully characterize a protein using an expression system such as Xenopus oocytes. When experimentally confirmed, a cDNA may be available as full length or overlapping EST (Expression Sequence Tag) clones as the result of Yugji Kohara's laboratory effort toward a systematic analysis of expression and function of C. elegans genome. For genes for which cDNA clones are not available, cDNA can be cloned relying on sequence information contained in highly conserved regions of the gene, relying on Caenorhabditis briggsae gene predictions or using non-biased methods based on RACE-PCR to identify extreme 3′ and 5′ ends (Bianchi et al., 2003).
Cloned cDNA must be inserted into a plasmid that ensures stability of the transcribed sequence and efficient translation into Xenopus oocytes. Vectors such as pSP64T (Promega), pGEM (Promega) and pCDNA3 (Invitrogen) all contain polyadenylation (poly(A)) sequences at the 3′ end of their cloning linker that render transcribed RNA stable and are suitable for oocytes expression (Huez et al., 1974). In addition plasmid pSP64T contains the 5′ and 3′ UTRs from Xenopus laevis β-globin. The β-globin sequences greatly enhance translation efficiency of heterologous mRNA transcripts in oocytes (Krieg and Melton, 1984). The vectors contain also RNA polymerase promoter sites (SP6 in pSP64T and T7 in PGEM and pCDNA3) that are used for the in vitro transcription.
In vivo most eukaryotic mRNAs have a 7-methyl guanosine cap structure at their 5′ end required for efficient message binding to ribosomes and efficient translation (Muthukrishnan et al., 1975). When in vitro transcribing RNA for oocyte expression studies, it is important to use a transcription kit that adds a guanosine cap to newly synthesized cRNA molecules. Failing to do so will dramatically reduce the expression of the protein of interest. mMESSAGE mMACHINE kits from Ambion contain a capped analog that is incorporated as the first G of the transcript. Ambion markets three kits containing three different RNA polymerases: SP6, T7 and T3. Depending on the promoter sequence of the vector employed and/or on the orientation of the cDNA, the appropriate kit to generate a sense transcript must be used.
DNA should be free of contaminating proteins and RNA. Most commercially available plasmid preparation systems yield DNA that is appropriate for in vitro RNA transcription using a transcription kit. Alternatively, DNA could be purified by standard phenol/chloroform/isoamyl alcohol extraction (Sambrook, 1989).
Circular plasmid templates generate long and heterogeneous RNA transcripts because RNA polymerases are highly processive. To produce a homogeneous population of transcripts, plasmid DNA should be linearized by digesting with a restriction enzyme that cuts at the 3′ end of the inserted cDNA. The restriction enzyme should be chosen with care. An enzyme that leaves 5′ end overhangs is optimal. Restriction enzymes leaving 3′ overhanging ends (for example Kpn I, Pst I, etc) are the least desirable because they yield low level of transcripts (Schenborn and Mierendorf, 1985).
(Use gloves throughout the procedure)
Digest plasmid DNA (5-10 μg) in a large volume reaction (150-200 μl) for at least 3 hours to ensure complete digestion of the template. Electrophorese 5 μl of the digestion mix to check for complete digestion.
Clean the digested DNA from restriction enzyme and restriction buffer either by phenol/chloroform/isoamyl alcohol extraction or using a DNA purification kit. PCR purification kit from QIAGEN is appropriate and recovers DNA at high yield. Elute or resuspend the DNA with 50 μl RNase-free water.
Ethanol precipitate DNA overnight at −20°C using 30 μl RNase-free 3 M sodium acetate and 1.2 ml 100% ethanol. Shorter precipitation times can be used but they may reduce the yield. Centrifuge 12,000g for 30 min at 4°C, wash once with 70% ethanol, decant supernatant completely and resuspend in 8 μl RNase-free water. Drying the pelleted DNA is not recommended since this may reduce its solubility and consequent transcription. Ethanol should be removed manually using a P20 pipette. Spinning the tube briefly helps collecting all the ethanol at the bottom. Make sure that all the ethanol is removed since it inhibits the transcription reaction.
Run 1 μl to check recovery of DNA and estimate its concentration by comparison with an appropriate size standard of known quantity or by measuring O.D.
Set up the transcription reaction following manufacture instructions and using 0.5–1.0 μg linear plasmid DNA. Incubate 1-2 h at 37°C. Incubation times may be longer for large transcripts.
Purify RNA either by standard ethanol precipitation (Sambrook, 1989) or using a commercially available RNA purification kit (for example RNAeasy from QIAGEN). Elute or resuspend the RNA in 25 μl RNase-free water, check for integrity and size by denaturing agarose gel electrophoresis (Sambrook, 1989). Estimate RNA concentration by spectrophotometry.
Oocytes are prepared from Xenopus laevis ovaries that can be either purchased from NASCO (www.enasco.com, Wisconsin) or extracted from adult females. Ovaries from NASCO are of high quality and represent a good alternative to culture and maintenance of Xenopus laevis in captivity, that bypasses surgeries and vertebrate animal protocol paper work. The protocol that follows includes the surgical procedure for extraction of the ovary.
Anesthetize an adult female by immersing it in 0.03% benzocaine in water. Make sure that the nostrils are above water. Place the frog ventral side up on crushed ice for approximately 20 min prior to, and throughout, the surgical procedure to maintain anesthesia.
Make a 1 cm incision in the lower abdominal wall near one leg to expose an ovary.
Carefully remove about one third of one ovary and place it in OR2 with CaCl2 or ND96 with CaCl2.
Suture the skin and the abdominal wall separately using 5-0 or 6-0 silk suture.
Allow the frog to recover from anesthesia in shallow water keeping nostrils above water.
Dissociate oocytes by collagenase treatment. Add 1ml of collagenase type II (2 mg/ml, in Ca2+ free ND-96 or OR-2) to 5 ml tubes and add approximately an equal volume of ovary in small pieces (~ 0.5 cm in diameter). Incubate for 1 h with gentle agitation at room temperature. Remove the collagenase solution and add fresh solution for another hour. Wash 4 times in fresh ND-96 or OR-2 with Ca2+ and incubate for a few hours to overnight prior to manual defolliculation. Removal of the follicular layer is actually easier several hours after collagenase treatment; in addition oocytes tend to be more fragile right after enzymatic treatment.
Manually defolliculate stage V and VI oocytes by forcing them through a Pasteur pipette with an opening slightly smaller than the diameter of the oocytes (smooth the opening by fire polishing). Incubate 3 to 4 hours prior to injection.
| OR2 solution (pH 7.2) | ND96 solution (pH 7.2) | ||
|---|---|---|---|
| NaCl | 82.5 mM | NaCl | 96 mM |
| KCl | 2.5 mM | KCl | 2 mM |
| Na2HPO4 | 1 mM | CaCl2 | 1.8 mM |
| Hepes | 5 mM | MgCl2 | 1 mM |
| MgCl2 | 1 mM | Hepes | 5 mM |
| CaCl2 | 1 mM | Gentamicin | 50 mg/l |
| Polyvinyl | 0.5 g/l | Pyruvic acid | 275 mg/l |
| Pyrolidone | 2.5 mM | Penicillin | 100 u/ml |
| Pyruvic acid | |||
| Penicillin | 100 u/ml | Streptomycin | 100 μg/ml |
| Streptomycin | 100 μg/ml | ||
Standard dissecting scope equipped with a cold light source (Figure 2a). The dissecting scope standard illumination system is not well suited for oocyte injection. Xenopus oocytes are not transparent and when illuminated from the bottom, are not clearly visible. In addition overheating caused by transmitted light may damage the oocytes.
|
Figure 2. Xenopus oocyte microinjection workstation. (a) The workstation shown here is based on the use of the Nanoject microinjector (Drummond Scientific Co., Broomall, PA, USA). Other types of injectors can be employed, for example the pressure driven microinjector Picospritzer(tm)II (General Valve Corp., Fairfield, NJ, USA). Labeled components are as follows: 1) dissecting microscope, 2) cold light source power supply, 3) cold light source goose-necks, 4) microinjector mounted on a coarse micromanipulator, and 5) switch pedal. (b) Oocyte injection chamber with microchip sockets for holding oocytes during injection.
Injector mounted on a micromanipulator (Figure 2a). For example Drummond Scientific markets Nanojet II specifically designed for oocyte injection. This injector is virtually vibration-free and can inject from 2.3 nanoliters to 69.0 nanoliters. The company also provides a micromanipulator, an adaptor, support bases and glass capillaries.
Oocyte chamber (Figure 2b). During injections, oocytes must be held in place. There is nothing on the market that can be used for this purpose, and therefore one has to be creative. Some researchers use a polypropylene mesh (0.8 mm grid) and others use indented Parafilm attached at the bottom of a Petri dish. We use microchip sockets salvaged from an old computer. The holes of microchip sockets are ~ 0.8 mm wide. Using this small home-made device also ensures that the oocytes picked are at the right developmental stage: less developed oocytes are smaller and go through the sockets.
Needle puller. P-87 and P-97 microelectrode pullers from Sutter Instruments are excellent and make very consistent needles of any shape and size, though a variety of other microelectrode pullers can be used.
(Use gloves throughout the procedure)
Pull microinjection needles. The Nanoject II requires the use of needles pulled from the glass provided by Drummond. Other injectors may require other types of capillaries. This information can be usually gathered from the injector's manufacturer.
"Backfill" the needles with oil (or other non-compressible fluid) before attachment to the injector. Silicone or mineral oil is frequently used. Backfilling is facilitated by using 30g x 2" needle and a syringe. Disposable spinal needles are also frequently used.
Ideally, the tip size should be 10-30 microns in size. Needle opening should be created by breaking off the tip end with forceps. This enables piercing the cell membrane much more easily with reduced tendency to become occluded with cellular material.
Force air out of the needle by pushing the "empty" button on the injector pedal. Air bubbles can affect the accuracy of the injection volumes.
Place at least 3 μl of RNA on a small piece of Parafilm and fill the needle by immersing the tip in the RNA solution drop.
Set the volume that you would like to inject (oocytes tolerate up to 69 nl well) and gently inject each oocyte by piercing the membrane and observing the oocyte "plump-up".
Incubate the oocytes in OR2 or ND96 with CaCl2 at 19°C or 20°C and change the solution daily.
Detailed information on electrophysiological techniques applicable to Xenopus oocytes can be found in two invaluable sources for electrophysiological methods: 1) "Ion channels of excitable membranes" by Bertil Hille (Hille, 2001) and 2) "Axon Guide: for electrophysiology and biophysics laboratory techniques" by Axon Instruments, Inc (free download at http://www.axon.com/mr_Axon_Guide.html) and in many excellent reviews (for example, Bezanilla and Stefani, 1998; Shih et al., 1998; Stefani, 1998; Stuhmer, 1998). In this section we will briefly review the two-electrode-voltage clamp (TEVC) technique as it is the most widely used electrophysiological technique to measure currents flowing through channels expressed in Xenopus oocytes. It should be noted though that execution of successful electrophysiological experiments requires more detailed description found elsewhere and hands-on training.
In TEVC, the oocyte membrane voltage is controlled (clamped) while currents flowing through channels or electrogenic transporters are measured. This is achieved by insertion of two electrodes through the oocyte membrane. One of the electrodes (the voltage-electrode) measures the membrane potential of the oocyte and connects to a feedback amplifier where this signal is compared with the command potential. The other electrode (the current-electrode) injects the current corresponding to the difference between the measured and the desired membrane potentials across the membrane and to the ground electrode. The electrodes are made of glass, are filled with a 3 M KCl solution and have an electric resistance between 0.2 and 3 MΩ. Other solutions with lower KCl concentrations and containing Ca2+ buffers such as EGTA can be used. Ca2+ buffers are used to inhibit endogenous Ca2+-activated Cl- currents (Kuruma and Hartzell, 1999). The electrodes are mounted on electrode-holders that contain a silver wire coated with a layer of AgCl2 which allows transmission of the signal from the KCl solution through the silver wire to the amplifier. The ground electrodes are made of 3 M KCl in 2-3% agar.
A stereomicroscope equipped with a cold light source, placed on a vibration isolation table under a Faraday cage (to shield from electric noise), is used to visualize the oocytes that are impaled using two coarse manipulators. Before impaling an oocyte, the voltage offset between electrodes and bath is cancelled. Oocyte impalement is done in current-clamp mode and successful penetration is confirmed by reading of the resting membrane potential on the amplifier display. A healthy non-injected oocyte has a membrane potential of ~ −40 mV, though expression of ion channels often induces deviations from this value. For example, expression of K+ selective channels drives the membrane potential to more negative values and expression of constitutively active Na+ or Ca2+ channels drives the resting potential to more positive values.
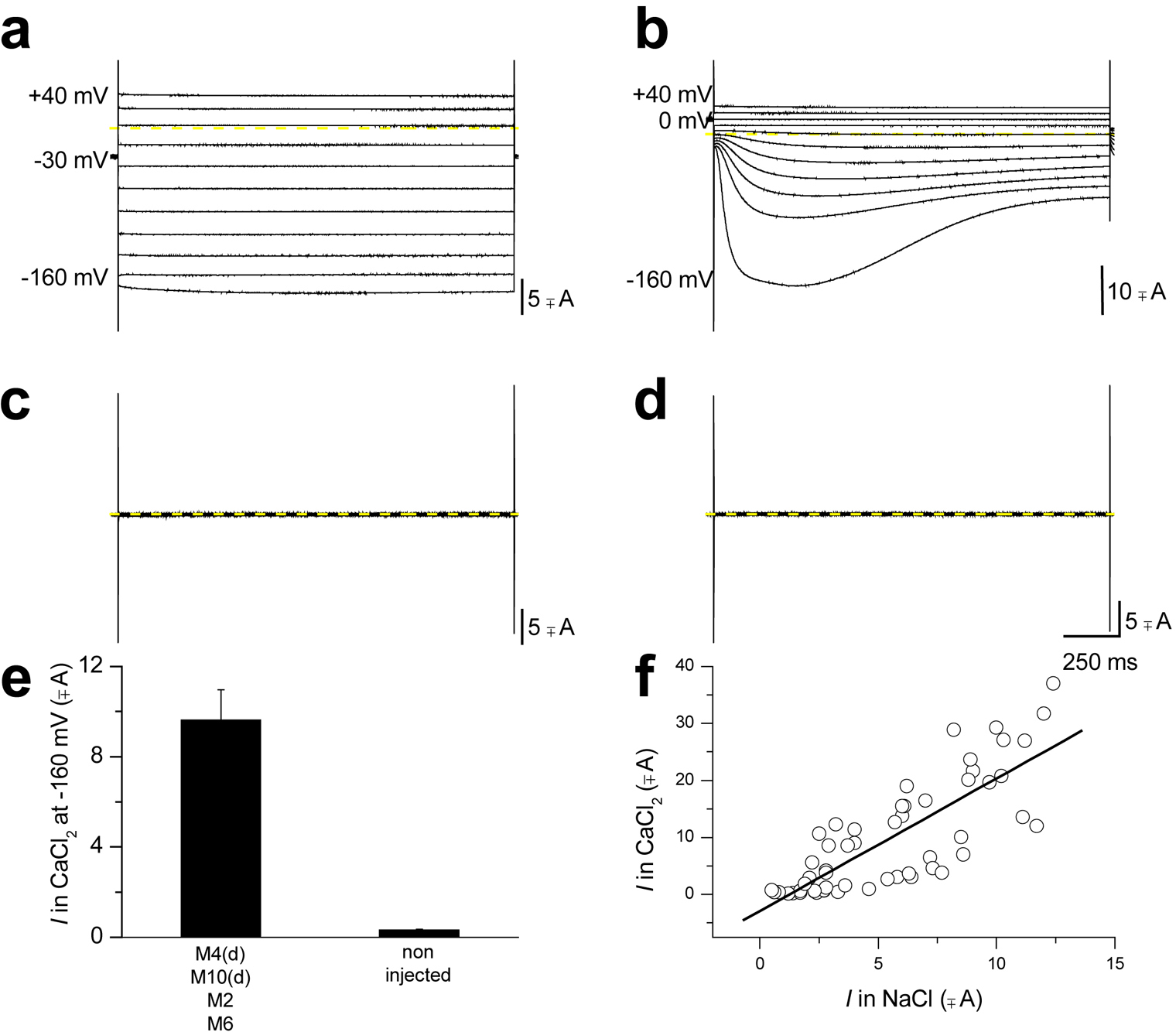
After both electrodes have been inserted, the amplifier is set in voltage-clamp mode and the voltage-clamp feedback gain, used to compensate for membrane capacitance, is increased to speed up the time response of the voltage clamp. Currents are then filtered (filtering frequency depends on the sampling rate, see the "Axon Guide" for more information) and recorded using an analog/digital converter and a computer. Examples of Na+ and Cl- currents recorded by TEVC from a Xenopus oocyte are shown in Figure 3.
|
Figure 3. Two electrode voltage-clamp recording of Xenopus oocytes expressing C. elegans DEG/ENaC toxic mutant channel MEC-4(d). (a) When an oocyte expressing MEC-4(d) (plus MEC-10(d), MEC-2 and MEC-6, designated as the MEC-4(d) channel complex) is exposed to a NaCl solution and is stimulated by voltages from −60 to + 40 mV (holding potential was −30 mV), it generates voltage-independent Na+ current. Conversely exposure of the same oocyte to a CaCl2 solution induces activation of a Xenopus oocytes endogenous Ca2+-activated Cl- current (b). (c) and (d) MEC-4(d) channel complex dependent Na+ currents and endogenous Cl- currents are not activated in non-injected oocytes. This implies that even the activation of the endogenous current is dependent on MEC-4(d) channel complex. (e) Average Cl- current at −160 mV recorded from oocytes injected with mec-4(d) channel complex and from non-injected oocytes; n = 56 and 16. Data are mean ± SE. (f) The amplitude of the Na+ current carried by MEC-4(d) channel complex is plotted against the amplitude of the Ca2+-activated Cl- current for each oocyte. Data points were fitted to a linear regression (slope = 2.3). Correlation between the amplitudes of the two currents again points towards dependence of one current upon the other. The endogenous Cl- current activated by exposure to Ca2+ only in oocytes injected with the mec-4(d) channel complex is activated by rise of intracellular Ca2+ (Kuruma and Hartzell, 1999) which is caused by entry of Ca2+ through MEC-4(d) (Bianchi et al., 2004). This is one case in which a Xenopus oocytes endogenous channel (Kuruma and Hartzell, 1999) was actually instrumental for discovering novel properties of an exogenous channel, namely MEC-4(d) permeability to Ca2+ ions, instead of being detrimental to electrophysiological recordings, (Bianchi et al., 2004). Modified from (Bianchi et al., 2004).
Detecting expressed proteins at the plasma membrane of Xenopus oocytes can be used to examine their trafficking to the membrane, their subcellular distribution or even interaction with other proteins (Shih et al., 1998). When drawing conclusions on protein trafficking based on observations made in Xenopus oocytes though, it should be kept in mind that oocytes are reared at 19°C–20°C while the organism from which the protein originates may have a different vital temperature. Studies on mammalian membrane proteins may be complicated by this problem (Delisle et al., 2004; Denning et al., 1992; Ficker et al., 2000; Kopito, 1999; Loo and Clarke, 1994), while for C. elegans proteins this factor may have less of an impact (nematodes can be reared at a wide range of temperatures from 15°C to 25°C). Nevertheless, it is important to keep in mind that certain mutant phenotypes are temperature-sensitive and a protein that is at the cell surface at 20°C may display a traffic defect at 25°C (Royal et al., 2005).
Membrane proteins can be visualized at the oocyte plasma membrane in two ways: 1) by tagging them with fluorescent proteins such as Green Fluorescent Protein (GFP) (Bianchi et al., 2004; Goodman et al., 2002; Bianchi et al., 1999), or 2) by immunostaining. There are at least three different protocols for oocyte immunostaining: two in which the oocytes are fixed and sliced prior to incubation with the antibodies (Chan et al., 1997; Mastroberardino et al., 1998) and one in which they are sliced after incubation with antibodies (Dent et al., 1989). For visualization of GFP-tagged proteins at the plasma membrane, oocytes must be fixed and sliced. Fixing and slicing methods described in protocol 1 are well-suited for visualization of GFP-tagged proteins.
(Adapted from Chan et al., 1997)
Examples of oocyte sections stained using this protocol are shown in Figure 4.
|
Figure 4. Immunostaining of C. elegans DEG/ENaC toxic mutant channel MEC-4(d) in Xenopus oocytes using a polyclonal antibody to MEC-4 (Lai et al., 1996). For this immunostaining protocol 1 was used (Chan et al., 1997). (a) Fluorescent photograph of an immunostained non-injected oocyte, showing no background fluorescence at the plasma membrane. (b) and (c) Oocytes expressing MEC-4(d) and MEC-4(A713T/ S726F), respectively, stained with anti-MEC-4 antibodies. In both cases accessory subunits MEC-10(d), MEC-2 and MEC-6 were coinjected along with a mutant form of primary pore-forming subunit MEC-4 (MEC-4(d)). Two second exposure time was used for all photographs. In this case oocyte immunostaining was used to determine whether lack of ionic currents associated with expression of double mutant channel MEC-4(d)/S726F was due to the channel being non-functional or not being correctly trafficked to the cell surface. Results of these experiments determined that the double mutant channel is at the cell surface but is not functional (modified from Bianchi et al., 2004).
Fix oocytes overnight at 4°C with 4% paraformaldehyde in PBS.
Wash three times for 5 minutes with PBS and imbed the oocytes in 3% low-melting point agarose in PBS. Pour enough warm (not hot) agarose in a 35 mm Petri dish to cover the bottom, drop 3 to 4 oocytes on the surface and cover with another layer of agarose.
Incubate the imbedded oocytes at 4°C for at least 2 hours to allow hardening of the agarose and then, using a vibrotome, section into 50 μm slices. Use PBS throughout the procedure for slice incubation. The agarose is used as a support and will be sliced as well, though in most cases the oocyte slice is released from the agarose soon after sectioning. Separation between agarose and oocyte slice can be forced by gentle pipetting with a fire-polished Pasteur pipette.
Block non-specific sites by incubation with 0.2% Bovine Serum Albumin (BSA) plus 0.1% Tween 20 in PBS overnight at 4°C or for 3–4 hours at room temperature. Non-fat dry milk is not used for oocyte immunocytochemistry because oocyte slices are white and are not visible in a milky solution. Transfer the slices from one solution to another using a fire-polished Pasteur pipette.
Incubate with the primary antibody in 1% Bovine Serum Albumin (BSA) plus 0.1% Tween 20 in PBS preferably over night at 4°C. A specific primary antibody gives the best results, though the protein of interest can be also tagged with an epitope (c-myc, Flag or HA) and visualized with commercially available antibodies (Mastroberardino et al., 1998).
Wash three times for 5 minutes with PBS and incubate with a fluorescent secondary antibody in 1% Bovine Serum Albumin (BSA) plus 0.1% Tween 20 in PBS for 1 hour at room temperature.
Wash three times for 5 minutes in PBS and mount the slices on glass slides using a medium appropriate for fluorescent microscopy (Vectorex from Vector for example).
Photograph slices using an inverted microscope equipped with 4x and 20x objectives. Microscopes typically used for C. elegans photography are well suited for oocyte photography.
(Adapted from Mastroberardino et al., 1998). Use this protocol to detect FLAG-tagged proteins.
Fix the oocytes with 3% paraformaldehyde in PBS for 4 h.
Place fixed oocytes on thin cork disks, embedded into cryo-embedding compound (Microm, Walldorf, Germany) and freeze them in liquid propane cooled by liquid nitrogen. Store the oocytes at −80°C until further use.
Section the oocytes in 6 μm slices using a cryostat and place them on chrom-alum gelatin-coated glass slides.
Use the tyramide signal amplification (TSA-Direct) kit (NEN, Boston, MA) according to the manufacturer's instructions. Incubate with anti-FLAG antibody diluted 1:100 in the TSA blocking buffer for 1 hour at room temperature.
Rinse slices with PBS containing 0.05% Tween and incubate with a 1:100 dilution of horseradish peroxidase-conjugated sheep anti-mouse Ig (1 hour at room temperature, Amersham, Arlington Heights, IL).
Wash with PBS-Tween and incubate with FITC-tyramide conjugates diluted 1:50 in the TSA diluent (1 hour at room temperature).
Wash slices in PBS-Tween and mount them in DAKO-Glycergel (Dako, Glostrup, Denmark) containing 2.5% of 1,4-diazabicyclo (2.2.2)-octane as a fading retardant (Sigma, St. Louis, MO).
(Adapted from Dent et al., 1989)
Remove the oocytes' vitelline membrane using fine tweezers.
Fix and permeabilize the oocytes in Dent's fixans (80% methanol, 20% DMSO) for 12 h at −20°C.
Wash with decreasing concentrations of ethanol and incubate with the primary antibody in 10% normal goat/donkey serum for 12 h at 4°C.
Wash with PBS and incubate with the secondary antibody for 1 h at room temperature.
Fix the oocytes again with 3.7% paraformaldehyde and dehydrate them with increasing concentrations of ethanol.
Imbed the oocytes into acrylamide (Technovit 7100 and 3040, Heraeus Kulzer, Germany) and when the acrylamide has dried, slice them in thin sections (5–6 μm) with a microtome.
Since immunoprecipitation and western blotting are standard techniques, in this section we will review only the sample preparation. The most important step when extracting proteins from Xenopus oocytes is to eliminate the large amount of yolk, which interferes with electrophoresis and immunodetection. Following are two protocols that allow elimination of the yolk and extraction of membrane proteins from Xenopus oocytes. The second protocol is designed for biotin labeling of membrane proteins and therefore allows separation of proteins that are at the cell surface from proteins that are retained in intracellular compartments.
(Adapted from Wible et al., 1998)
Harvest 20–30 oocytes and resuspend them in 0.3 M sucrose plus 10 mM sodium phosphate (pH 7.4) containing a protease inhibitor mixture (for example Complete from Roche).
Homogenize with 20 strokes in a glass homogenizer on ice and spin the samples at 3,000 g for 10 min at 4°C. Recover the supernatant and discard the pellet (nuclei and debris). When recovering the supernatant use extra care to avoid transferring of the yolk which is usually floating at the surface.
Spin the supernatant at 48,000 g for 1 h at 4°C to pellet membranes.
Resuspend the pelleted membranes in 0.3 M sucrose plus 10 mM sodium phosphate (pH 7.4) containing a protease inhibitor mixture and subject to SDS-PAGE. The samples should be mixed with reducing SDS sample buffer (7% SDS) and heated at 90°C for 15 min before separation on SDS-PAGE.
(Adapted from Muller et al., 1993)
Procedure for biotin labeling of oocytes membrane proteins.
Wash 50-75 oocytes five times in OR-2 solution and place them into an agarose-coated Petri dish filled with 2 ml of OR-2.
Carefully add the fresh membrane-impermeant reagent NHS-LC-biotin stock (2 mg/500 μl of OR-2) to the dish. After 10 min at room temperature stop the reaction by adding 1 ml of 500 mM glycine, pH 7.4.
Wash the oocytes three times in 500 mM glycine, pH 7.4, followed by two washes in OR-2.
Transfer the oocytes to a microcentrifuge tube and add 0.5 ml of lysis buffer (2% Nonidet P-40, 150 mM NaCl, 2 mM CaCl2, 20 mM Tris, pH 7.4, plus 2 μM leupeptin and pepstatin and 1 mM phenylmethylsulfonyl fluoride).
Pass the oocytes 20 times through a 200-μl pipette, and centrifuge the homogenate twice (1,000 × g, 10 min, 4°C) to remove the yolk. Sonicate the supernatant for 1 min and then centrifuge again at 1,000 × g (10 min, 4°C).
Dialyze the supernatant overnight at 4°C against streptavidin buffer (SAv buffer) (0.3% Nonidet P-40, 500 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM Tris, pH 8, plus 2 μM leupeptin and pepstatin and 1 mM phenylmethylsulfonyl fluoride) and then centrifuge for 30 min at 14,000 rpm at 4°C to remove insoluble material.
Incubate the supernatant overnight with 75 μl of streptavidin-agarose bead suspension (previously washed three times in SAv buffer).
Separate supernatant and pellet by low speed (2,000 rpm) centrifugation.
Precipitate the supernatant with 5% trichloroacetic acid, resuspend in 150 μl of Laemmli sample buffer, boil and electrophorese.
When using Xenopus oocytes for heterologous expression of proteins, several considerations should be kept in mind. As mentioned above, Xenopus oocytes are kept best at 19°C or 20°C, therefore mutant mammalian channels or transporters whose trafficking is defective only at restricted higher temperatures (typically above 35°C) may display a normal phenotype when expressed in frog oocytes (Delisle et al., 2004; Denning et al., 1992; Ficker et al., 2000; Kopito, 1999; Loo and Clarke, 1994). C. elegans proteins may not suffer this problem, but this consideration should be kept in mind when working with temperature-sensitive mutant proteins (Royal et al., 2005).
Frog oocytes express endogenous channels and transporters at a low level. These endogenous proteins can interfere with analysis of expressed proteins in at least two ways: 1) they can be upregulated by expression of exogenous proteins and interfere with the analysis of the protein of interest, and 2) they can form heteromultimers with the proteins translated from injected RNA. One example for the formation of heteromultimeric ion channels is the IKs channel. The IKs K+ channel is formed by two types of subunits, KCNE1 (or minK) and KCNQ1 (or KvLQT1). When KCNE1 was expressed alone in Xenopus oocytes it induced IKs channels and therefore was thought to form channels by itself (Hausdorff et al., 1991). Cloning of KvLQT1 revealed that the IKs channel in Xenopus oocytes is formed by the exogenously expressed KCNE1 and an endogenous KCNQ1 isoform (xKCNQ1) (Sanguinetti et al., 1996). Sometimes oocyte endogenous currents are critical tools for deciphering properties of exogenous channels. For example, the oocyte endogenous Ca2+-activated Cl- current (Kuruma and Hartzell, 1999) was instrumental for expression-cloning the ionotropic glutamate receptor iGluR3 (Boulter et al., 1990) and for discovering C. elegans toxic DEG/ENaC channel MEC-4(d) permeability to Ca2+ ions (Figure 3; Bianchi et al., 2004).
Finally cellular environment, particularly signaling pathways, may differ from that of the cell where the ion channel of interest is normally expressed, calling for extreme caution when studying channel modulation by secondary messengers in oocytes. In addition there are differences between C. elegans and vertebrates in the types of sugars attached during glycosylation of membrane proteins (Schachter, 2004) and glycosylation affects function and trafficking of ion channels (Dietrich et al., 2003; Khanna et al., 2001; Petrecca et al., 1999; Schwalbe et al., 1995). C. elegans membrane proteins contain a predominance of oligomannose sugars identical to those found in vertebrates. The more complex and hybrid N-glycans that are highly abundant in vertebrates are either absent in C. elegans or present at undetectable levels.
Xenopus laevis oocytes constitute a simple, low-cost and well-controlled system for expression of exogenous proteins in large quantity. This expression system is particularly amenable to the study of membrane proteins using electrophysiological techniques because of the large size and spherical shape of the oocyte, both of which facilitate insertion of electrodes and good voltage control. Other techniques applied to Xenopus oocytes allow detection and isolation of membrane proteins using fluorescence and immunochemical tags. These techniques can be used to complement electrophysiological recordings for a thorough analysis of a channel or transporter, in its wild type or mutant forms. Despite the limitations of oocytes (endogenous channels and transporters, and rearing temperature) they are still a powerful method for studying several aspects of ion channel biology, including permeation properties, structure-function relationships, regulation by intracellular and extracellular molecules and stoichiometry. Researchers interested in C. elegans membrane proteins have used Xenopus oocytes to produce data that significantly advanced our understanding of worm channels, transporters and receptors function and regulation.
The authors are grateful to Drs. Itzhak Mano and Alfred L. George Jr. for critical reading of the manuscript. Research support from NIH grants R21-NS049511 to L. B. and R01-NS37955 to M. D. is acknowledged.
Bezanilla, F., and Stefani, E. (1998). Gating currents. Methods Enzymol. 293, 331–352. Abstract
Bianchi, L., Gerstbrein, B., Frokjaer-Jensen, C., Royal, D.C., Mukherjee, G., Royal, M.A., Xue, J., Schafer, W.R., and Driscoll, M. (2004). The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat. Neurosci. 7, 1337–1344. Abstract Article
Bianchi, L., Kwok, S.M., Driscoll, M., and Sesti, F. (2003). A potassium channel-MiRP complex controls neurosensory function in Caenorhabditis elegans. J. Biol. Chem. 278, 12415–12424. Abstract Article
Bianchi, L., Shen, Z., Dennis, A.T., Priori, S.G., Napolitano, C., Ronchetti, E., Bryskin, R., Schwartz, P.J., and Brown, A.M. (1999). Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum. Mol. Genet. 8, 1499–1507. Abstract Article
Boulter, J., Hollmann, M., O'Shea-Greenfield, A., Hartley, M., Deneris, E., Maron, C., and Heinemann, S. (1990) Molecular cloning and functional expression of glutamate receptor subunit genes. Science 249, 1033–1037. Abstract
Browne, C.L., and Werner, W. (1984). Intercellular junctions between the follicle cells and oocytes of Xenopus laevis. J. Exp. Zool. 230, 105–113. Abstract Article
Chan, K.W., Sui, J.L., Vivaudou, M., and Logothetis, D.E. (1997). Specific regions of heteromeric subunits involved in enhancement of G protein-gated K+ channel activity. J. Biol. Chem. 272, 6548–6555. Abstract Article
Delisle, B.P., Anson, B.D., Rajamani, S., and January, C.T. (2004). Biology of cardiac arrhythmias: ion channel protein trafficking. Circ. Res. 94, 1418–1428. Abstract Article
Denning, G.M., Anderson, M.P., Amara, J.F., Marshall, J., Smith, A.E., and Welsh, M.J. (1992). Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761–764. Abstract Article
Dent, J.A., Polson, A.G., and Klymkowsky, M.W. (1989). A whole-mount immunocytochemical analysis of the expression of the intermediate filament protein vimentin in Xenopus. Development 105, 61–74. Abstract
Dietrich, A., Mederos y Schnitzler, M., Emmel, J., Kalwa, H., Hofmann, T., and Gudermann, T. (2003). N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J. Biol. Chem. 278, 47842–47852. Abstract Article
Ficker, E., Thomas, D., Viswanathan, P.C., Dennis, A.T., Priori, S.G., Napolitano, C., Memmi, M., Wible, B.A., Kaufman, E.S., Iyengar, S., et al. (2000). Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome. Am. J. Physiol. Heart Circ. Physiol. 279, H1748–H1756. Abstract
Goodman, M.B., Ernstrom, G.G., Chelur, D.S., O'Hagan, R., Yao, C.A., and Chalfie, M. (2002). MEC-2 regulates C. elegans DEG/ENaC channels needed for mechanosensation. Nature 415, 1039–1042. Abstract Article
Gurdon, J.B. (1973). The translation of messenger RNA injected in living oocytes of Xenopus laevis. Acta. Endocrinol. Suppl. (Copenh) 180, 225–243. Abstract
Gurdon, J.B., Lane, C.D., Woodland, H.R., and Marbaix, G. (1971). Use of frog eggs and oocytes for the study of messenger RNA and its translation in living cells. Nature 233, 177–182. Abstract Article
Gurdon, J.B., Lingrel, J.B., and Marbaix, G. (1973). Message stability in injected frog oocytes: long life of mammalian alpha and beta globin messages. J. Mol. Biol. 80, 539–551. Article
Gurdon, J.B., Woodland, H.R., and Lingrel, J.B. (1974). The translation of mammalian globin mRNA injected into fertilized eggs of Xenopus laevis I. Message stability in development. Dev. Biol. 39, 125–133. Abstract
Hausdorff, S.F., Goldstein, S.A., Rushin, E.E., and Miller, C. (1991). Functional characterization of a minimal K+ channel expressed from a synthetic gene. Biochemistry 30, 3341–3346. Abstract Article
Hille, B. (2001). Ion Channels of Excitable Membranes, 3rd ed., Sunderland, Ma: Sinauer Associates, Inc.
Huez, G., Marbaix, G., Hubert, E., Leclercq, M., Nudel, U., Soreq, H., Salomon, R., Lebleu, B., Revel, M., and Littauer, U.Z. (1974). Proceedings: Role of the polyadenylic segment in the translation of globin messenger RNA. Hoppe-Seyler's Z. Physiol. Chem. 355, 1211. Abstract
Khanna, R., Myers, M.P., Laine, M., and Papazian, D.M. (2001). Glycosylation increases potassium channel stability and surface expression in mammalian cells. J. Biol. Chem. 276, 34028–34034. Abstract Article
Kopito, R.R. (1999). Biosynthesis and degradation of CFTR. Physiol. Rev. 79, S167–S173. Abstract
Krieg, P.A., and Melton, D.A. (1984). Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 12, 7057–7070. Abstract
Kuruma, A., and Hartzell, H.C. (1999). Dynamics of calcium regulation of chloride currents in Xenopus oocytes. Am. J. Physiol. 276, C161–C175. Abstract
Lai, C.C., Hong, K., Kinnell, M., Chalfie, M., and Driscoll, M. (1996). Sequence and transmembrane topology of MEC-4, an ion channel subunit required for mechanotransduction in Caenorhabditis elegans. J. Cell. Biol. 133, 1071–1081. Abstract Article
Laskey, R.A., and Gurdon, J.B. (1973). Induction of polyoma DNA synthesis by injection into frog-egg cytoplasm. Eur. J. Biochem. 37, 467–471. Abstract Article
Laskey, R.A., and Gurdon, J.B. (1974). The translation of viral RNAs in frog oocytes. Hamatol. Bluttransfus. 14, 272–274. Abstract
Laskey, R.A., Gurdon, J.B., and Crawford, L.V. (1972). Translation of encephalomyocarditis viral RNA in oocytes of Xenopus laevis. Proc. Natl. Acad. Sci. U.S.A. 69, 3665–3669. Abstract Article
Loo, T.W., and Clarke, D.M. (1994). Prolonged association of temperature-sensitive mutants of human P-glycoprotein with calnexin during biogenesis. J. Biol. Chem. 269, 28683–28689. Abstract
Mastroberardino, L., Spindler, B., Forster, I., Loffing, J., Assandri, R., May, A., and Verrey, F. (1998). Ras pathway activates epithelial Na+ channel and decreases its surface expression in Xenopus oocytes. Mol. Biol. Cell. 9, 3417–3427. Abstract
Miledi, R., Parker, I., and Sumikawa, K. (1982a). Properties of acetylcholine receptors translated by cat muscle mRNA in Xenopus oocytes. EMBO J. 1, 1307–1312. Abstract
Miledi, R., Parker, I., and Sumikawa, K. (1982b). Synthesis of chick brain GABA receptors by frog oocytes. Proc. R. Soc. Lond. B. Biol. Sci. 216, 509–515. Abstract
Miledi, R., and Woodward, R.M. (1989a). Membrane currents elicited by prostaglandins, atrial natriuretic factor and oxytocin in follicle-enclosed Xenopus oocytes. J. Physiol. 416, 623–643. Abstract
Miledi, R., and Woodward, R.M. (1989b). Effects of defolliculation on membrane current responses of Xenopus oocytes. J. Physiol. 416, 601–621. Abstract
Muller, A.H., Gawantka, V., Ding, X., and Hausen, P. (1993). Maturation induced internalization of beta 1-integrin by Xenopus oocytes and formation of the maternal integrin pool. Mech. Dev. 42, 77–88. Abstract Article
Muthukrishnan, S., Both, G.W., Furuichi, Y., and Shatkin, A.J. (1975). 5'-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 255, 33–37. Abstract Article
Petrecca, K., Atanasiu, R., Akhavan, A., and Shrier, A. (1999). N-linked glycosylation sites determine HERG channel surface membrane expression. J. Physiol. 515, 41–48. Abstract Article
Royal D.C., Bianchi L., Royal M.A., Lizzio, M., Jr., Mukherjee G., Nunez Y.O., and Driscoll, M. (2005). Temperature-sensitive mutant of the Caenorhabditis elegans neurotoxic MEC-4(d) DEG/ENaC channel identifies a site required for trafficking or surface maintenance. J. Biol. Chem. 280, 41976–41986. Abstract Article
Sanguinetti, M.C., Curran, M.E., Zou, A., Shen, J., Spector, P.S., Atkinson, D.L., and Keating, M.T. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83. Abstract Article
Schachter, H. (2004). Protein glycosylation lessons from Caenorhabditis elegans. Curr. Opin. Struct. Biol. 14, 607–616. Abstract Article
Schenborn, E.T., and Mierendorf, R.C., Jr. (1985). A novel transcription property of SP6 and T7 RNA polymerases: dependence on template structure. Nucleic Acids Res. 13, 6223–6236. Abstract
Schwalbe, R.A., Wang, Z., Wible, B.A., and Brown, A.M. (1995). Potassium channel structure and function as reported by a single glycosylation sequon. J. Biol. Chem. 270, 15336–15340. Abstract Article
Shih, T.M., Smith, R.D., Toro, L. and Goldin, A.L. (1998). High-level expression and detection of ion channels in Xenopus oocytes. Meth. Enzymol. 293, 529–556. Abstract
Stefani, E.a.B.F. (1998). Cut-open oocyte voltage-clamp technique. Meth. Enzymol. 293, 300–318. Abstract
Stuhmer, W. (1998). Electrophysiological recordings from Xenopus oocytes. Meth. Enzymol. 293, 280–318. Abstract
Tapper, A.R. and Goerge, A.L. Jr. (2003). Heterologous expression of ion channels. Methods Mol. Biol. 217, 285–94. Abstract
Theodoulou, F.L., and Miller, A.J. (1995). Xenopus oocytes as a heterologous expression system. Methods Mol. Biol. 49, 317–340. Abstract
Wang, H.C., Beer, B., Sassano, D., Blume, A.J., and Ziai, M.R. (1991). Gene expression in Xenopus oocytes. Int. J. Biochem. 23, 271–276. Abstract Article
Wible, B.A., Yang, Q., Kuryshev, Y.A., Accili, E.A., and Brown, A.M. (1998). Cloning and expression of a novel K+ channel regulatory protein, KChAP. J. Biol. Chem. 273, 11745–11751. Abstract Article
Wickens, M.P., Woo, S., O'Malley, B.W., and Gurdon, J.B. (1980). Expression of a chicken chromosomal ovalbumin gene injected into frog oocyte nuclei. Nature 285, 628–634. Abstract Article
Woodland, H.R., Gurdon, J.B., and Lingrel, J.B. (1974). The translation of mammalian globin mRNA injected into fertilized eggs of Xenopus laevis. II. The distribution of globin synthesis in different tissues. Dev. Biol. 39, 134–140. Abstract
*Edited by William Schafer. WormMethods editor, Victor Ambros. Last revised May 30, 2006. Published August 1, 2006. This chapter should be cited as: Bianchi, L. and Driscoll, M. Heterologous expression of C. elegans ion channels in Xenopus oocytes (August 1, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.117.1, http://www.wormbook.org.
Copyright: © 2006 Laura Bianchi and Monica Driscoll. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. Phone: 732-445 7183 or 87, Fax: 732-445 7192, E-mail: bianchi@biology.rutgers.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.