Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Because of their free-living life cycle alternatives, Strongyloides and related nematode parasites may represent the best models for translating C. elegans science to the study of nematode parasitism. S. stercoralis, a significant pathogen of humans, can be maintained in laboratory dogs and gerbils. Biosafety precautions necessary for work with S. stercoralis, though unfamiliar to many C. elegans researchers, are straightforward and easily accomplished. Although specialized methods are necessary for large-scale culture of the free-living stages of S. stercoralis, small-scale cultures for experimental purposes may be undertaken using minor modifications of standard C. elegans methods. Similarly, the morphological similarities between C. elegans and the free-living stages of S. stercoralis allow investigational methods such as laser cell ablation and DNA transformation by gonadal microinjection to be easily adapted from C. elegans to S. stercoralis. Comparative studies employing these methods have yielded new insights into the neuronal control of the infective process in parasites and its similarity to regulation of dauer development in C. elegans. Furthermore, we have developed a practical method for transient transformation of S. stercoralis with vector constructs having various tissue- and cell-specific expression patterns and have assembled these into a modular vector kit for distribution to the community.
The breadth and depth of C. elegans science, exemplified by WormBook, represents an under-utilized resource for the study of parasitic nematodes. Conversely, the global health impact of nematode parasitism (Hotez et al., 2006) provides a strong but seldom cited rationale for research on C. elegans. The recent addition of chapters on parasitic nematodes to WormBook (see Trichinella spiralis; Strongyloides spp.) represents an effort to attract the attention of parasitologists to this resource and to interest the C. elegans community in the potential applications of their work to an important group of pathogens. Of all the parasitic nematodes, Strongyloides and relatives, by virtue of their free-living life cycle alternatives, appear best suited to the immediate application of methodology from C. elegans. Free-living larvae, males and females of Strongyloides sp. (see Figure 1) and Parastrongyloides sp., resemble C. elegans in their feeding habits and in many aspects of their overall body plans (see Strongyloides spp.). Like C. elegans, the free-living stages of these parasites may be grown or, in the case of Parastrongyloides trichosuri, propagated on agar plates with bacterial lawns (Grant et al., 2006a; Schad, 1989; Viney, 1999). As will be discussed below, our group has exploited anatomical similarities between C. elegans and Strongyloides stercoralis to adapt not only standard culture methods but also techniques for neuronal ablation and gene delivery in C. elegans to this parasite. Grant and colleagues have similarly achieved transgenesis and developed other basic components of a genetic system in P. trichosuri (Grant et al., 2006a; Grant et al., 2006b; Newton-Howes et al., 2006).
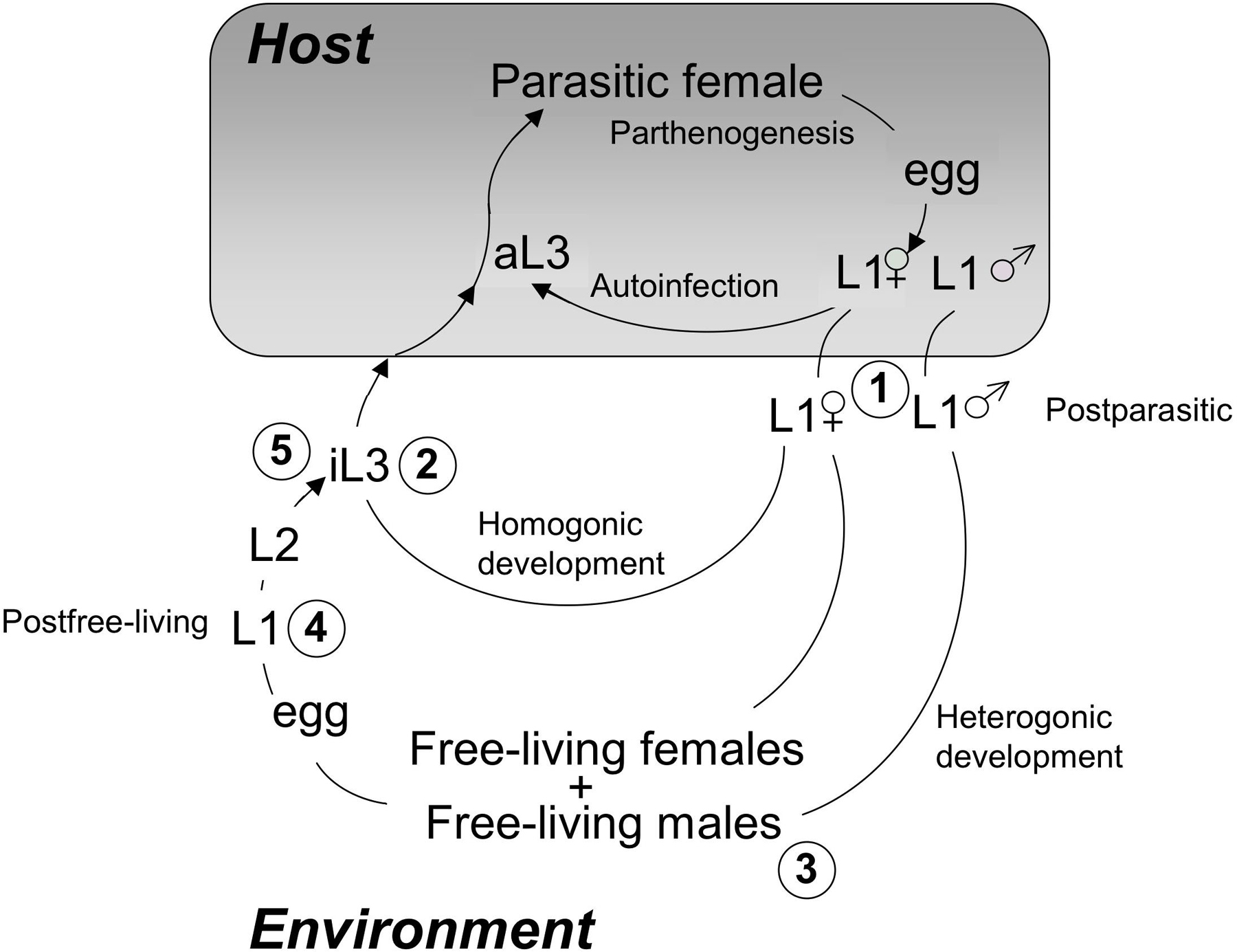
All progeny of free-living S. stercoralis adults are fated to become iL3, which must infect a host in order to develop further (see Figure 1). Consequently, worms of this species are capable of undergoing only one generation of development outside the host and continuous laboratory rearing requires maintenance of host animals. S. stercoralis is a parasite of primates (including humans) and dogs (Schad, 1989). We maintain the strain of S. stercoralis designated UPD (University of Pennsylvania, Dog) as a subclinical infection in purpose-bred, mix-breed laboratory dogs. We typically, infect dogs by inoculating them subcutaneously with 3000 infective third-stage larvae (iL3).
|
Figure 1. Life cycle of S. stercoralis. The diagram depicts the three possible developmental fates of hatchling first-stage larvae (L1 within the shaded box indicating the host). First, is the autoinfective cycle, unique to S. stercoralis, in which development from the L1 to the autoinfective L3 (aL3) occurs within the gut of the primary host. Autoinfection can lead to fulminant cycles of development and a highly pathogenic disseminated infection. All postparasitic male L1 develop to free-living adult males. Postparasitic female L1 passed in the feces may undergo development by either of two alternative pathways. The homogonic cycle involves direct development to the infective L3 (iL3), and heterogonic development involves development to the free-living female and, following mating, production of a generation of free-living progeny. All progeny of the free-living adults develop to the iL3. Larval stages between the post-parasitic L1 and the aL3, iL3 or free-living adult as well as between the iL3 or aL3 and the parasitic female are omitted for the sake of clarity. Encircled numbers refer to stages that may be recovered using the culture protocols indicated in Table 1.
Their ability to tolerate chronic infection with larger numbers of adult worms and their higher output of fecal larvae make dogs the most practical hosts for laboratory maintenance of S. stercoralis. However, the use of such a species for experiments requiring many replicate host infections or the maintenance of numerous parasite strains may be prohibitively expensive and a source of ethical concern. While attempts to infect rats and mice with S. stercoralis have failed, gerbils, Meriones unquiculatus, have been found to be susceptible to both infection and autoinfection (see Figure 1, "aL3") by this parasite. The factors that promote autoinfection in the natural canine or human hosts, immunological immaturity or corticosteroid treatment, do so in the gerbil as well, making this small mammal a valuable model in which to study host-parasite interactions that govern the autoinfective process (Nolan et al., 1993; Kerlin et al., 1995; Nolan and Schad, 1996; Nolan et al., 1999).
The iL3 of S. stercoralis invade the host percutaneously. Convention holds that they migrate via the microvasculature to the lungs where they break out of the capillary bed, enter the alveoli and then proceed to the intestine via the trachea. Work by Schad and colleagues (Schad et al., 1989), however, suggests that the majority of larvae may actually migrate via a variety of other somatic tissues. In any case, the larvae then undergo two molts to the parasitic female, which invades crypts of the intestinal mucosa. The primary symptom of uncomplicated human strongyloidiasis is abdominal pain, mimicking that associated with peptic ulcer, and acute infections are marked by the presence of first stage larvae in fresh fecal specimens and by eosinophilia. As indicated above, S. stercoralis is unique among the parasitic nematodes in its capacity to undertake autoinfection, and immunosuppressed individuals should refrain from working with active cultures of this parasite.
With regard to safety of animal husbandry staff, it is important to reinforce that 1.) S. stercoralis iL3 infect percutaneously, 2.) larvae found in feces in the first 24 hours (L1) are not infectious and 3.) 24 hours at normal room temperature are required for L1 passed in the feces to reach the infective stage via the homogonic route (see Figure 1). Therefore, provided a schedule of daily, thorough cage cleaning is maintained (standard operating procedure in most institutional vivaria), the risk of transmission should be minimal. Staff should wear protective clothing consisting at minimum of disposable gloves, gowns, shoe covers and caps, as well as eye and face protection when cleaning cages. In general, these will not constitute special precautions for maintenance of S. stercoralis, but rather standard procedure for laboratory animal husbandry staff at most institutional laboratory animal operations.
As with animal husbandry operations, laboratory safety measures mandated at most institutions are adequate to protect against infection with S. stercoralis. Although the presence of infective larvae may be predicted with some precision based on the age of the culture and the incubation temperature (Table 1), it must be borne in mind that at least a small proportion of post-parasitic L1 in most S. stercoralis strains develop directly to the iL3 within 24-48 hours of deposition (ie. the homogonic route). Since the exact deposition time of fecal samples is usually uncertain, it is therefore good practice to regard all S. stercoralis cultures, regardless of age as potentially containing infectious parasites. All disposable culture waste should be accumulated in clearly marked biohazard bags and autoclaved prior to discard. Non-disposable glassware, Baermann funnel components (see Protocol 2 below) and other implements contaminated with free-living larvae and adults of S. stercoralis may be decontaminated with a 20% solution of a commercial surgical scrub such as ChlorhexiDerm (2% Chlorhexidine, DVM Pharmaceuticals, Miami, Florida). Surgical scrub should be added to Baermann funnel effluent waste at a similar concentration to kill free-living larvae and adults prior to disposal. Because of their highly resistant cuticles, iL3 are impervious to Chlorhexidine and other conventional surgical scrubs even after several hours of exposure. Implements and solutions containing iL3 can be chemically decontaminated with Lugol's Iodine (10 mg/ml KI + 10 mg/ml I2 in distilled water).
The challenge in designing preparative culture methods for free-living larvae and adults of S. stercoralis is to simulate the complex nutritional milieu of moist, fecally contaminated soil while preventing anaerobia. The charcoal coproculture method described below (see Protocol 1) accomplishes this and, assuming adequate output of fecal larvae by the host, provides excellent yields of worms. Methods for isolation of S. stercoralis and related parasites from such a chemically and microbiologically complex environment (e.g., the Baermann funnel technique, Protocol 2) generally exploit the natural migratory behaviors of the worms, as do some methods for further purification (the agarose migration technique, Protocol 3). Otherwise, by virtue of their similarity to other soil-dwelling nematodes, many standard cultures methods may be adapted from C. elegans methodology for small-scale analytical culture.
Table 1. Incubation protocols for derivation of selected life stages of S. stercoralis from charcoal coproculture.
| Developmental stage | Figure 1 reference** | Incubation protocol | |
|---|---|---|---|
| Temperature (°C) | Incubation time | ||
| Postparasitic L1 | 1 | Available immediately in freshly deposited feces | |
| Directly developing (homogonic) iL3 | 2 | 22 | 24 hr |
| Free-living adults | 3 | 20 | 72 hr |
| 22 | 48 hr | ||
| Postfree-living L1* | 4 | 23.5 | 48 hr |
| iL3 | 5 | 22 | 3-5 days |
| * Old free-living adults will also be present in these cultures of postfree-living L1. The L1 may be manually selected from Baermann effluent using flame constricted Pasteur pipets and a dissecting microscope. | |||
| **Figure 1 reference numbers indicate where specified life stages occur in the S. stercoralis life cycle in Figure 1 (see encircled numerical labels on key life stages). | |||
Low temperature incubator(s). One or more incubators capable of regulating temperature in the range of 4-37°C are essential for cultivating the free-living stages of S. stercoralis. A single unit set for 26°C can be made to suffice for many purposes. However, we find it advantageous to have three incubators set at 20°, 22° and 25° to allow maximum flexibility in planning experiments and incubating selected stages at optimum temperatures.
Fume hood. Feces of S. stercoralis-infected dogs present no hazards of exposure to infective larvae via the contact aerosol inhalant routes during the culture procedure outlined below. However, the aesthetic drawbacks of working with fresh canine feces on an open laboratory bench are easy to imagine. Happily, once this material is dispersed on activated charcoal as called for in this protocol, it is virtually odor-free. However, prior to this step, odors may be offensive. As a measure against this, we carry out the culture preparation procedure (4.2.3, steps 2–6) in a fume hood. A small biohazard bag for discard of waste materials, such as soiled sample bags and tongue depressors used for fecal collection, should be kept in the fume hood and changed frequently.
A standard dissecting microscope
Disposable nitrile gloves
Wooden tongue depressors to collect fecal samples.
Self-closing plastic sample bags to contain fecal samples.
Small Tupperware box to transport bagged samples from the vivarium.
Plastic Petri dishes, disposable 100 mm
Filter paper circles, Whatman #1 (or equivalent), 100 mm diameter.
Absorbent paper bench liner
Plastic beaker, Nalgene, 1 liter capacity.
Biohazard bag (12′ X 24″) with stand
Plastic containers with lids to contain coprocultures in incubators.
Activated bone charcoal, (5 X 8 mesh). This should be autoclaved and washed thoroughly with tap water prior to use. Washing can be carried out by transferring about 1 kg of the material to a suitably sized Styrofoam shipping container with one or more small holes in a bottom corner. Place the box with charcoal in the sink and fill with tap water. Allow the tap water to drain and repeat. The washed charcoal should be allowed to drain overnight and may then be stored covered in this container next to (or inside) the fume hood.
Deionized (DI) water in a plastic squeeze bottle.
Large funnel(s), 150 mm diameter, glass or Nalgene plastic.
Tubing, rubber or Tygon plastic, with diameter gauged to give a watertight fit over stem of large funnel (#13).
Pinch clamps, thumbscrew operated.
As with laboratory safety precautions (see Section 3.3), minimum standards for protective clothing required for entry into well regulated institutional vivaria will be adequate for safe handling of S. stercoralis infected dogs and their feces.
Obtain fresh feces from an infected dog. Feces, which should be formed and free of urine contamination, can be scooped from the cage bottom using a wooden tongue depressor and placed in a self-sealing plastic sample collection bag. To attain some uniformity in the age of fecal parasite larvae, it is best to fix a standard time of day to do fecal collections. We typically do this between 09:00 and 09:30 hrs, prior to scheduled cage cleaning by lab animal husbandry personnel.
Place sample bag with feces in a small Tupperware container for transport from the vivarium to the laboratory. Handle this box with a gloved hand.
Mix 1 part feces with 3 parts activated charcoal and 1 part DI water in a 1-liter plastic beaker. Estimate relative volumes feces and charcoal by eye.
Mix thoroughly with a tongue depressor.
Add more water if necessary. Charcoal grains should appear moist, and uniformly coated with fecal slurry, but there should not be excess water apparent in the mixture.
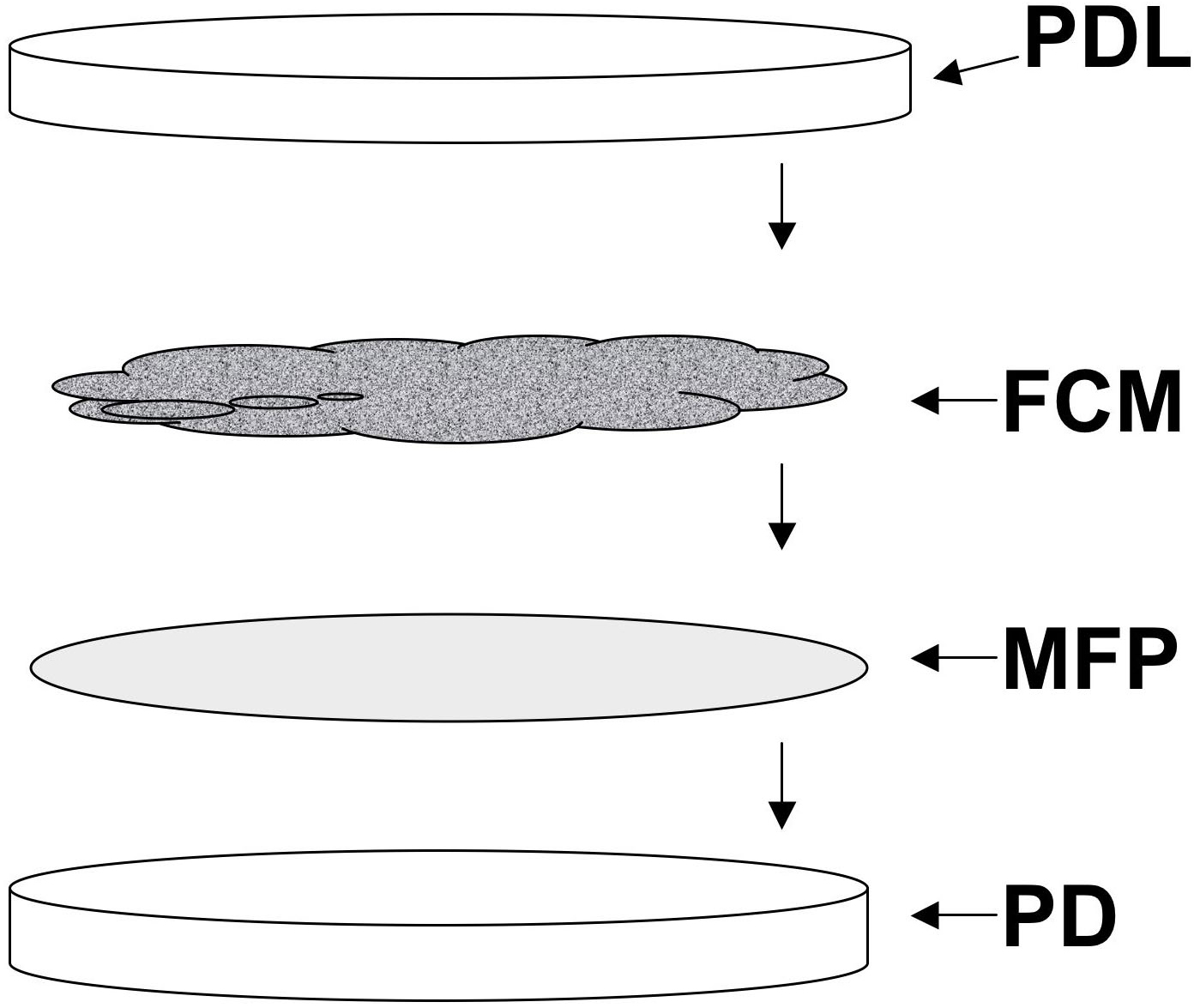
Line several 100 mm Petri dishes with Whatman #1 filter paper circles (see Figure 2). Moisten these to the point of glistening with DI water.
Transfer fecal charcoal mixture to each dish and cover (see Figure 2). Dishes should be full, but the mixture should not touch the lid. Do not seal the plates.
|
Figure 2. Components of a charcoal coproculture of Strongyloides stercoralis. A 100 mm Petri dish lined with a moist Whatman #1 filter paper circle (MFP). FCM, fecal-charcoal mixture consisting of one part infected canine feces and one part de-ionized water evenly dispersed on 4 parts activated granular bone charcoal. The moist filter paper lined Petri dish is filled to the brim, but not above it, with the fecal charcoal mixture and covered with the plate lid (PDL), labeled with the date and other essential data.
Incubate at predetermined temperature (22-27°C, depending on experimental procedures) at ~75% R.H. This humidity may be achieved by placing dishes on a Styrofoam flat floating in approximately 0.5 in. of water in a Tupperware crisper box.
Some common incubation protocols and their results are given in Table 1.
The Baermann funnel is commonly used to isolate small parasitic nematodes from solid materials such as feces, soil or animal tissue. It exploits the tendency of these worms to migrate from a solid into a surrounding liquid medium when stimulated by slightly elevated temperature and then to settle to the substratum (Bowman, 1995). By using an ordinary laboratory funnel to contain the incubating sample, and taking some simple filtration measures, it is possible to collect and concentrate large numbers of worms in small volumes of medium, relatively free of fecal debris.
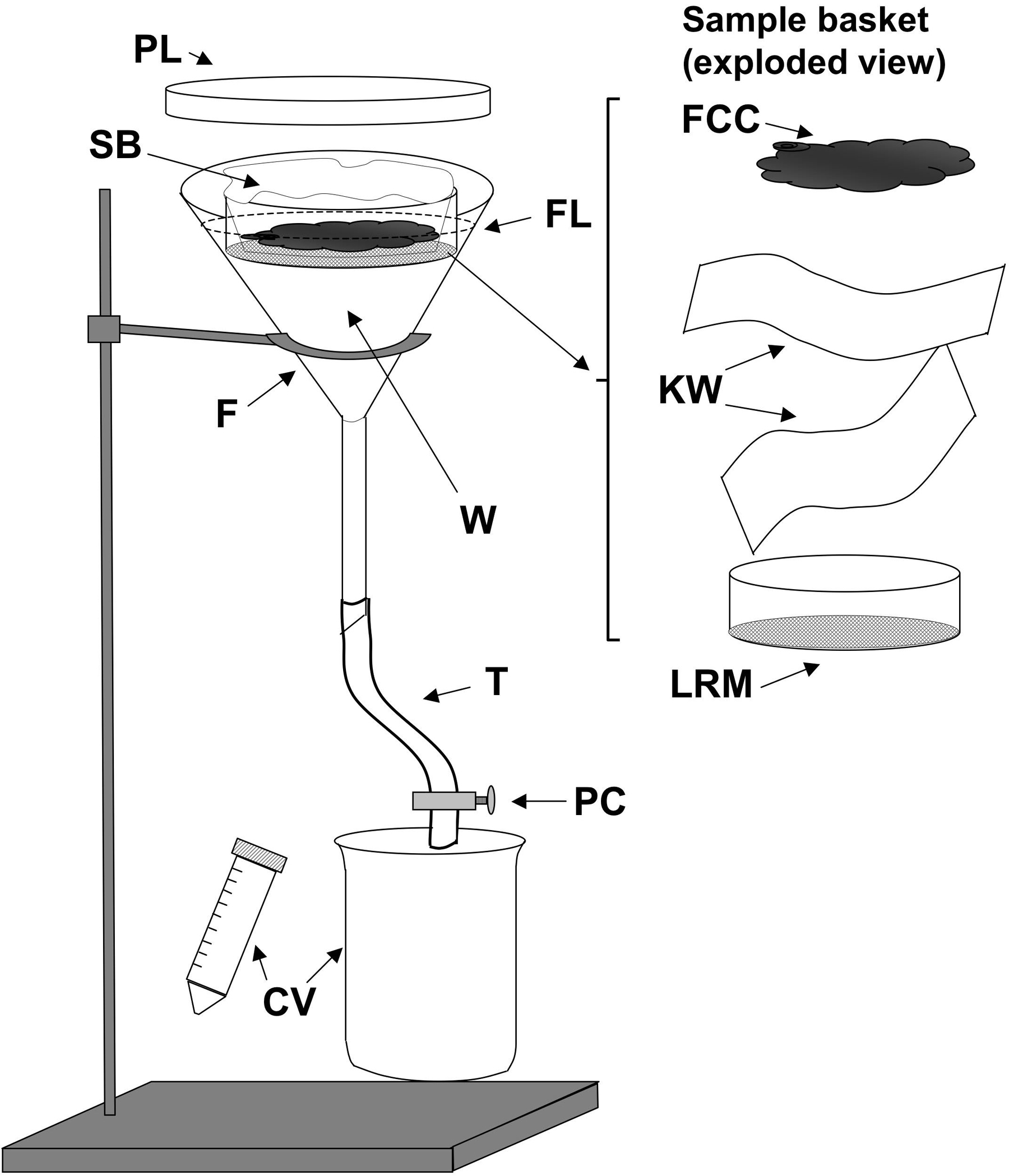
Assemble the Baermann funnel apparatus as shown in Figure 3. We find that Tygon plastic food delivery tubing (0.5 inch inside diameter, Fisher Scientific Cat. No. 14-169-21D) because it is transparent, has the advantage of allowing visual detection of substantial numbers of worms accumulating at the bottom of the apparatus, giving some indication of parasite yield prior to harvest. The disadvantage of Tygon tubing of this size is that due to its thick walls, a very tight closure using a thumbscrew operated clamp is required to achieve a watertight seal. The lid from a 150 mm disposable plastic Petri dish fits well over the top of the funnel. When properly cleaned between procedures, all of these components may be reused many times.
Line the sample basket, basically a sieve consisting of a Lucite ring with coarse nylon mesh (bridal netting, available at fabric stores) attached with cement, with two layers of laboratory tissue (see Figure 3). A pair of Kimwipes oriented at 90° angles works well for this purpose. A somewhat cruder, but no less effective sample basket can be made by cutting the bottoms from two half pint-sized plastic food containers and nesting one tightly inside the other with a single layer of bridal netting between.
Select one or two charcoal coprocultures of the desired age based on Table 1.
Use a tongue depressor to place the contents of the plate(s) including the filter paper liners on top of the Kimwipes lining the sample basket.
Carefully place the sample basket with culture contents into the funnel.
Close the tubing clamp tightly and then gently pour tap water at 43°C down the side of the funnel until the level rises just above the culture contents. Use the tongue depressor to gently submerge the edges of the tissue liner into the water. Note: The roughly 600–800 ml of tap water needed may be warmed to 43°C in a microwave oven or achieved by mixing hot and cold water at the tap.
|
Figure 3. A Baermann apparatus as used to isolate Strongyloides stercoralis stages from coprocultures. F, 150mm glass or plastic funnel; W, tap water at 43°C; FL (dashed line) fluid level in funnel; SB, Sample basket shown to right in exploded view (FCC, fecal charcoal culture contents; KW, two layers of Kimwipes or other laboratory tissue; LRM Lucite ring with nylon mesh affixed with cement; T, rubber or plastic tubing; PC, pinch clamp; CV, catch vessels, conical centrifuge tube, or beaker.
Cover the funnel with the Petri dish lid and incubate for 1-3 hours. As a safety measure in the event of leakage, we place a one-liter plastic beaker underneath each funnel. To avoid hypoxic stress to the isolated parasites we harvest and plate the proceeds of Baermann funnels at one-hour intervals.
Harvest the worms accumulated in the funnel stem by carefully loosening the thumbscrew of the tubing clamp and releasing 15-20 ml of fluid into a 50 ml conical centrifuge tube. If desired, the yield may be determined by transferring 20μl aliquots of the collected worm suspension onto a microscope slide, counting under the dissecting microscope. The total number of parasites is then calculated based on the volume of the suspension, which can be approximated from the graduations on the centrifuge tube). Note that S. stercoralis settle rapidly, and the worms must be re-suspended prior to taking each aliquot.
Allow the worms to sediment at 1 x g for approximately 15 min; collect with a long Pasteur pipet, and transfer to an appropriate receptacle. For worms to be used in active cultures, transfer the sedimented with approximately 100μl of fluid to a standard NGM agar plate spread with E. coli OP50 (see Maintenance of C. elegans; Lewis and Fleming, 1995), and place the plate on the bench with its lid ajar for drying. Drying in a laminar flow hood accelerates this process and decreases exposure to airborne contaminants.
For applications requiring observation and analysis of smaller populations of worms, such as screening of phenotypes or manual selection of genetic transformants, the free-living stages of S. stercoralis may be cultured on standard NGM/OP50 plates (see Maintenance of C. elegans; Lewis and Fleming, 1995). Since the worms used to inoculate such cultures will be derived from the septic environment of a coproculture, significant carryover of fecal bacteria onto the NGM/OP50 plate is likely. Contamination due to bacteria and fungi external to the worms may be minimized, but not eliminated by either of two approaches: allowing the worms to migrate through a semi-solid matrix of low gelling temperature agar (see Protocol 3) or subjecting them to density gradient centrifugation (see Protocol 4). Since the worms carry fecal bacteria within their guts, neither of these procedures will result in a sterile preparation. To date we have been unscuccessful at adapting the standard alkaline sodium hypochlorite technique used for axenization of C. elegans eggs (see Maintenance of C. elegans; Lewis and Fleming, 1995) to yield viable S. stercoralis eggs in quantity.
Prepare 10 ml of 3% low melting point agarose (Sigma Alderich Cat. No. A9414) in BU saline (Hawdon and Schad, 1991). Boil in the microwave oven for 30-40 sec on high power.
BU saline (1 liter):
| 50 mM Na2HPO4 | 7.098 g |
| 22 mM KH2PO4 | 2.99 g |
| 70 mM NaCl | 4.09 g |
| H2O | to 1 liter |
Mix molten agarose thoroughly and place in a 37°C water bath until temperature equilibrates.
Similarly, place a concentrated suspension of S. stercoralis to be purified (e.g., the sediment from a Baermann funnel isolation reduced to 2-3 ml volume) as well as a 20 ml aliquot of BU saline into the 37°C water bath to equilibrate.
Thoroughly mix 1 volume of low melting point agarose at 37°C with 2 volumes of concentrated parasite suspension to give a 1:3 dilution to a final concentration of 1% low melting point agarose. Note: since low melting point agarose solidifies rapidly at temperatures below 37°C, we typically perform this operation with tubes in the water bath.
Place a 100 mm Petri dish(s) on ice, and carefully pipet 5 ml of parasite suspension in 1% agarose into the center of the plate. Try to form bead of agarose that does not contact the sides of the plate. To facilitate this step, a level cooling platform may be made by embedding an inverted aluminum block insert from a laboratory dry bath in crushed ice.
Allow the dish with agar bead to rest on ice for a few minutes until completely solidified.
Add 10–20 ml of warmed BU saline to the plate with solidified agar. Cover the plate and incubate at 37°C for 30 min. Viable worms should migrate from the agar matrix into the surrounding medium during this period.
Without disturbing the solid agar bead, pipet the liquid medium from the incubated plate and transfer to a 15 ml conical centrifuge tube. Worms may be allowed to sediment by gravity (approx. 15 min) or by centrifugation at 500 rpm for 5 min.
Wash worms two times in M9 buffer
During final wash spin, add 10 ml of 40% Percoll solution to a 15 ml conical centrifuge tube.
40% Percoll solution 100 ml:
| Percoll | 40 ml |
| HEPES | 0.596 g (25mM) |
| Penicillin/Streptomycin stock soln.* | 1 ml |
| H2O | To 100 ml |
*10,000 U/ml Penicillin, 10 mg/ml Streptomycin
Remove supernatant from washed worm pellet and re-suspend worms in the residual volume.
Use a Pasteur pipet to carefully layer the suspended pellet onto the Percoll solution.
Spin at 1000 rpm for 2 min. Repeat spin as necessary to pellet the worms. Because of their ability to swim upwards, iL3 may require more than one spin.
Remove Percoll supernatant by aspiration.
Subject worm pellet to two washes with M9 buffer.
Transfer worms in final wash pellet to a clean OP50 plate.
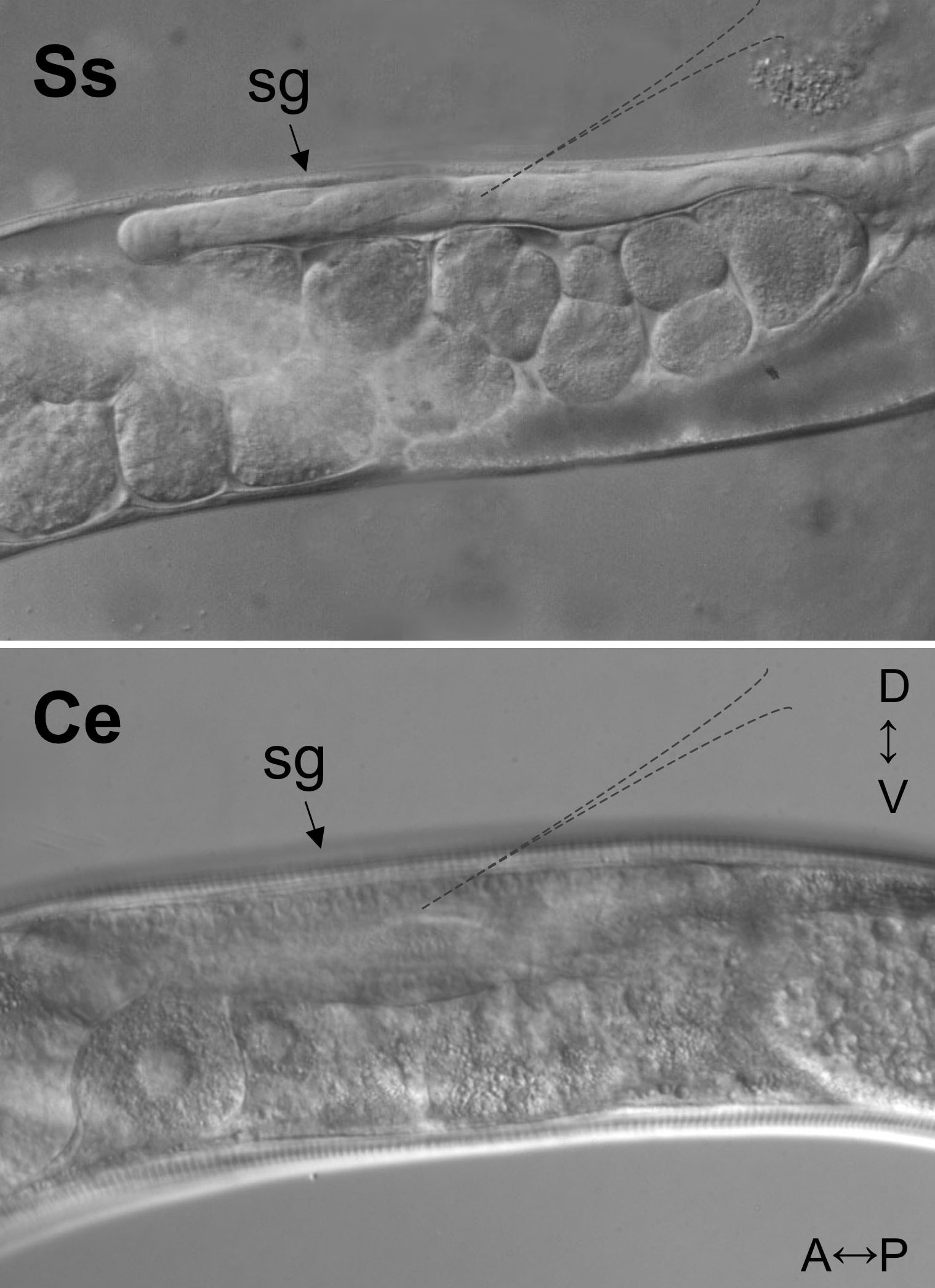
The size, transparency and body plan of a free-living S. stercoralis female is so similar to that of a C. elegans hermaphrodite that published techniques for delivery of DNA constructs into C. elegans by gonadal microinjection (see Transformation and microinjection; Mello and Fire, 1995) may be used for this parasite virtually without modification (Li et al., 2006; Lok and Massey, 2002). Persons experienced with gonadal microinjection of C. elegans will succeed almost immediately with S. stercoralis but will notice two relatively minor differences in the two worms. First, the reproductive tracts of free-living S. stercoralis females and C. elegans hermaphrodites are highly similar, both being in the form of a double recurved tube with the vulva placed at mid-body. However, as pictured in Figure 4, the syncytial ovary of S. stercoralis, corresponding to the usual point of injection in C. elegans, is generally narrower and less transparent than that of C. elegans, and individual oocyte nuclei are less visible by DIC microscopy. Second, due perhaps to relative fragility of its cuticle or to a higher internal hydrostatic pressure, S. stercoralis free-living females are somewhat more susceptible than C. elegans to injury by injection of an excessive fluid volume or by large or improperly placed needles. The most frequent manifestation of this is prolapse of the gonad from the injection wound. This problem may be overcome by using needles with an outside tip diameter no greater than 5μm and by keeping the injection pressure under 20 pounds per square inch.
|
Figure 4. Differential interference contrast (DIC) images of syncytial gonads (sg) of the S. stercoralis free-living female (Ss) and the C. elegans hermaphrodite (Ce). A↔P and D↔V indicate anterior-posterior and dorsal-ventral directions, respectively, for specimens in both panels. Dashed outlines in each panel suggest the optimum point of needle placement for microinjection. Note that nuclei in the syncytial gonad of C. elegans are clearly visible and not so in the less transparent gonad of S. stercoralis.
Early attempts to transform S. stercoralis involved a series of constructs in which various C. elegans and S. stercoralis promoters were fused to the coding sequence of gfp in standard C. elegans vectors from the vector kit maintained previously by A. Fire and colleagues (Stanford University). These constructs incorporated either of two commonly used C. elegans 3′ UTRs, that of unc-54 or of let-858. Invariably, transgene constructs incorporating C. elegans 3′ UTRs were expressed in apparently unregulated fashion in degenerating embryos of S. stercoralis and never in normally developing F1 larvae (Li et al., 2006; Lok and Massey, 2002). Regulated, cell- and tissue-specific transgene expression was not achieved until both 5′ and 3′ regulatory sequences derived from the parasite were incorporated. For example, as with all such constructs, gfp expression under the control of the S. stercoralis era-1 promoter in a standard C. elegans vector background (pPD 95.81) is always of the unregulated type described above. In contrast to this, substitution of the endogenous Ss era-1 3′ UTR for the C. elegans unc-54 3′ UTR in the original vector is sufficient to bring about tissue-specific expression in normally developing F1 larvae (Li et al., 2006). Sequence comparison reveals that the endogenous Ss era-1 3′ UTR, which gives regulated expression, contains three canonical polyadenylation signals whereas the previously used C. elegans unc-54 element, which gives unregulated expression, contains only one (Li et al., 2006). This signal alone may be insufficient to properly terminate message transcription in S. stercoralis. The Ss era-1 3′ UTR is also sufficient to give regulated expression of gene constructs incorporating other S. stercoralis promoters (see Table 2). In combination with this element, the promoter from the cellular actin gene Ss act-2 gives expression primarily in body wall muscle of F1 larvae while that of the Gα protein-encoding gene Ss gpa-3 gives a pattern of expression in sensory neurons remarkably similar to that of its C. elegans ortholog (Jansen et al., 1999). Our group has developed a "toolkit" of S. stercoralis vectors for distribution, which are designed to accept promoters in all reading frames upstream of the gfpS65C coding sequence with artificial introns and the Ss era-1 3′ UTR. This toolkit is available from the author upon request.
Table 2. Tissue- and cell-specific expression of three promoter:gfp fusion constructs incorporating the Ss era-1 3′ UTR in F1 transformant larvae of S. stercoralis.
| Vector | Promoter | Reporter | 3′ UTR | Expression pattern |
|---|---|---|---|---|
| pPV230.13 | Ss era-1 | gfpS65C | Ss era-1 | Intestinal cells |
| pAJ08 | Ss act-2 | Body wall muscle | ||
| pAJ09 | Ss gpa-3 | 8 amphidial, 2 phasmidial neurons |
For experiments involving smaller numbers of worms, we typically cultivate microinjected free-living female worms along with 1-2 males per female, and their F1 progeny on standard NGM agar plates spread with OP50 bacteria. We have been able to rear isolated P0 transformant females (plus males) and their broods in this manner (Li et al., 2006) as well as pools of up to 30 transformant females and an equal number of males. Carryover of fecal bacteria to the NGM/OP50 plates may be problematic and can be minimized by washing free-living adults from Baermann isolations with two changes of M9 buffer.
For larger cohorts of P0 transformants (100-200 individuals), as would be required for host passage of transgenics, we temporarily pool females as they are microinjected, along with a roughly equal number of males, on NGM/OP50 plates. At the end of a day's injection session, microinjected females and males are washed from the plate with a small volume of M9 buffer and transferred as a pool to a charcoal coproculture dish (see section 4.2.3) made with feces from a non-infected dog. These plates are incubated at 22°C for 5-6 days to allow development to the iL3, which are collected by the Baermann funnel protocol (see section 4.3).
Behavioral or selectable transformation markers have not been developed as yet for S. stercoralis. However, gfp expression under the control of strong promoters such as that of Ss act-2 constitutes a workable visual marker for transformation. Ss act-2p::gfp transformants are easily hand-selected under a stereomicroscope equipped with epifluorescence.
Although reliable protocols for derivation of stable transgenic lines of S. stercoralis are not yet in hand, we have used experimental infections in gerbils to accomplish the following required steps in this process. First, we have shown that relatively small cohorts of transgenic S. stercoralis (i.e., inocula containing 100-200 F1 transformant iL3) can establish patent infections in gerbils and that construct DNA can be maintained in transgenic populations through two host passages with intervening free-living generations in culture to the F5 generation (Li et al., 2006). This initial finding was made with a non-expressed construct, the C. elegans-based pTG96_2, which contains the sur-5 promoter fused to gfp and the unc-54 3′ UTR (Gu et al., 1998), and so host passages had to be carried out without selection. More recent attempts to effect host passage of F1 larvae showing regulated expression of the Strongyloides-based Ss act-2p::gfp construct, pAJ08, have yielded gfp-expressing parasitic females. However no gfp-expressing individuals have been observed among the progeny of these transgenic parasitic females and, thus, we have not yet succeeded in establishing a stable gfp-expressing line.
The tendency of gonadal microinjection to result in the incorporation of transgenes into multi-copy episomal arrays has led to some interest in biolistic DNA transfer as an alternate method of gene delivery facilitating the isolation of stable lines of C. elegans with low-copy chromosomal integrations or homologous gene replacements. Microparticle bombardment has also been used for transient transfection of the adult filariae such as Litomosoides carinii (Jackstadt et al., 1999) and Brugia malayi (Higazi et al., 2002). One of the major impediments to the use of biolistics for heritable transgenesis in parasitic nematodes is the low frequency of germline transformation by this method relative to the number of adult or L4 parasites that may be practicably obtained. The most efficient systems for biolistic transformation of C. elegans allow derivation of one stable transformant line per 20,000 individuals bombarded, and typical bombardments involve 104-105 animals (Praitis et al., 2001). The ease of large-scale C. elegans culture and the availability of powerful, selectable co-transformation markers such as the unc-119 mutant rescue make it relatively straightforward to amass the numbers of worms required for bombardment and to select integrated lines (see Transformation and microinjection). It has been possible for us to prepare cohorts of 5-7 x 104 free-living S. stercoralis adults using the fecal output of two infected dogs and scaled up versions of the charcoal coproculture and Baermann isolation techniques described above (see Protocols 1 and 2). Initial problems with overgrowth of these large cultures by fecal bacteria can be overcome using the percoll gradient method (see Protocol 4) to decontaminate the worms. However, using the PDS/1000 He bombardment apparatus we have been unable, thus far, to detect F1 transformants expressing the Ss act-2p::gfp construct, pAJ08 (Table 2), which has proven so effective in producing F1 transformants by microinjection. A selectable transformation marker, possibly one encoding resistance to an anthelmintic drug, would greatly facilitate the detection of transformants in large populations of bombarded S. stercoralis, and our future efforts in this area will concentrate on this approach.
Microsurgical ablation of cells and observation of resulting developmental and behavioral deficits is a standard approach to the study of cell function in whole organisms. C. elegans' transparent cuticle and the fact that few of its cells are strictly required for survival make microlaser ablation, particularly of neurons, an important part of the methodological repertoire for this model organism (Bargmann and Avery, 1995). The power of this method was recently demonstrated in the elucidation of a complete “.....behavioral circuit for navigation in C. elegans, from sensory input to motor output...” (Gray et al., 2005). As has been the case for microinjection of transgene DNA, we have been able to exploit morphological similarities between C. elegans and S. stercoralis to adapt established techniques for laser cell killing from the model organism to the parasite and have used this method to investigate the neuronal control of various behaviors and developmental events associated with the infective process (Ashton et al., 2007). The method for cell ablation in S. stercoralis detailed in Protocol 5 below is a modification of that reported for C. elegans (Bargmann and Avery, 1995).
Prepare 20 ml of 1.5% low gelling temperature (low melting point) agarose (Type 1-A, Sigma Aldrich Chemical Co., St. Louis, Missouri) in BU saline (see Section 4.5, Protocol 3) and divide into 2 ml. aliquots. These may be stored in the refrigerator in sterile snap-cap tubes.
Melt one aliquot of 1.5 low melting point agarose; add 6μl of the anesthetic 1-phenoxy 2-propanol (Janssen Chemica, Geel, Belgium), and vortex.
Use molten agarose with anesthetic to prepare mounting pads on microscope slides, using one layer of laboratory tape to gauge pad thickness, as described (see Methods in cell biology; Bargmann and Avery, 1995)
Use a finely drawn capillary tube to transfer post-parasitic first-stage larvae, 5-8 per slide, to the anesthetic pad. These larvae are should be freshly isolated from feces or charcoal coproculture as described in 4.2. The 1-2μl of fluid transferred with the larvae will allow the worms to undulate a few times before coming to rest on the pad, thereby orienting themselves laterally.
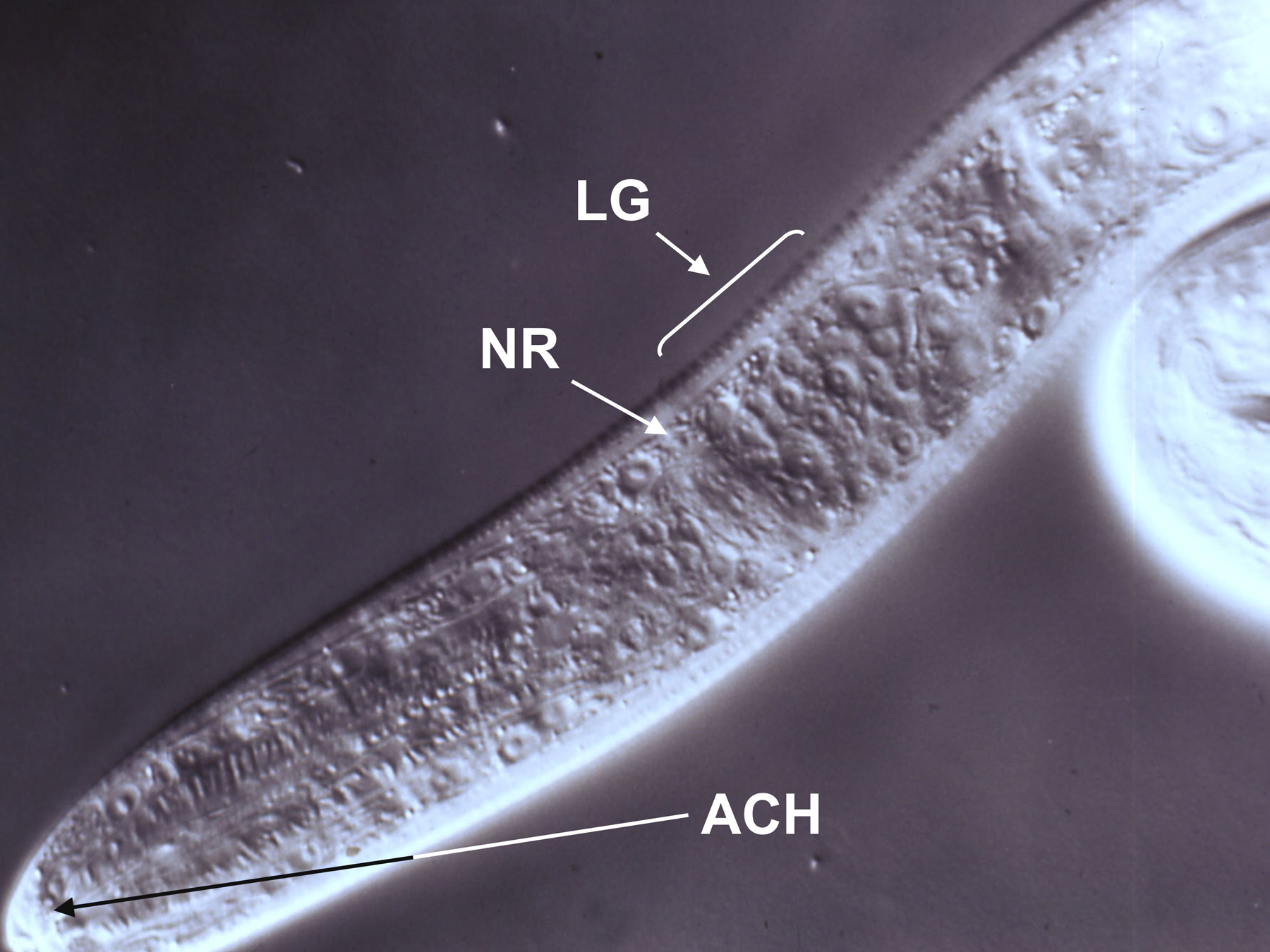
View anesthetized larvae at 1000X magnification by differential interference contrast microscopy and identify the cell bodies, which should be clearly visible at this magnification (see Figure 5), by comparison to a published map of the cell bodies of the L1. (Ashton et al., 1998).
|
Figure 5. DIC image of a first-stage S. stercoralis larva positioned for microlaser ablation of amphidial cell bodies. Cell bodies within the lateral ganglion (LG) are clearly visible just posterior to the nerve ring (NR). Dendritic processes of the amphidial cells converge in the amphidial channel (ACH). Following ablation of the target amphidial cell in this focal plane, the microscope and laser would be refocused on the other cell body in the neuron pair and this cell ablated as well. Image courtesy of G. A. Schad.
Ablate the target cell in the upper focal plane using 50-60 pulses of the laser. Amphidial neurons are paired and bilateral. If the worms are oriented laterally, it should be possible to refocus the microscope and laser directly downward through the organism to the cell body of the underlying member of the neuron pair for ablation.
Following ablation, apply a lethal laser burst to kill any unoperated larvae. This will prevent their being confused with successfully operated worms later in the experiment.
Carefully transfer operated larvae from the anesthetic pad to a dish containing 1.5 ml of PBS for recovery. Once the operated worms resume normal motility, transfer them via micropipet to the center well of a 15 X 60 mm organ culture dish containing 1 ml of 1% NGM agar to which a small patch of canine fecal solids (approximately 1 mm diameter) has been applied with the tip of a wooden applicator stick.
Depending on the phenotype of interest, worms may be observed immediately or cultured to the appropriate stage. Our group's interest is primarily in neuronal control of switching between homogonic and heterogonic developmental alternatives and of resumption of development by iL3, so we typically screen larvae for phenotypes following incubation for 24-30 hr at 20° C.
The recent successes with transgenesis in strongyloidoid parasites (Grant et al., 2006b; Li et al., 2006) are highly encouraging. However, in the cases of both Strongyloides and Parastrongyloides, some key components of a practical transgenesis system have yet to be identified. We have succeeded in designing a number of reporter constructs S. stercoralis based upon cell- and tissue-specific promoters linked to the in vivo reporters gfp and mrfp. However, it remains for us to establish any of these in a stable transformant line through host passage, and much of our current effort is directed toward this goal. By contrast, stable transgenic lines of P. trichosuri expressing constructs incorporating the (β-GAL reporter system have been established (Grant et al., 2006b), but, to date, constructs that will express gfp or other in vivo reporters have not been reported for this parasite.
The utility of RNA interference (RNAi) as a method of investigating gene function in C. elegans is well substantiated (Fire et al., 1998; Johnson et al., 2005; Tabara et al., 1998; Tavernarakis et al., 2000; Timmons et al., 2001). Robust RNAi effects following natural uptake or electroporation of dsRNA have been observed in the filariae (Aboobaker and Blaxter, 2003; Ford et al., 2005; Hashmi et al., 2004) and the trichostrongyles (Hussein et al., 2002; Issa et al., 2005), parasites occupying Clades III and V, respectively, in the contemporary nematode phylogeny (Blaxter et al., 1998). By contrast, despite concerted efforts, none of the laboratories investigating Strongyloides or Parastrongyloides, members of nematode Clade IV, have been able to demonstrate clear RNAi effects in these parasites following application of dsRNA in various configurations via the common routes of administration. Our own efforts have shown a significant knock down of mRNA encoding the S. stercoralis homolog of unc-54 in the progeny of free-living females microinjected with homologous dsRNA. However, whereas unc-54 shows a strong paralyzed RNAi phenotype in C. elegans, no such phenotype has been seen in S. stercoralis. Among planned approaches to address this problem in the future are combinatorial experiments in which we will attempt to enhance RNAi effects in S. stercoralis by knocking down mRNAs encoding putative orthologs of molecules such as LIN-53, which modulate RNAi effects in C. elegans, simultaneously with targets of interest.
The extensive body of work by Viney and co-workers on S. ratti demonstrates that limited forward genetic approaches may be feasible with the strongyloidoid parasites. For example, they have shown that clonal populations, varying in key developmental traits, may be selected and maintained through host passage, that genetic exchange occurs during mating in the free-living generation (Viney et al., 1993) and that free living populations may be chemically mutagenized, screened for phenotypes of interest and selected for relevant mutations (Viney et al., 2002). The ability of P. trichosuri to undergo multiple free-living generations in culture make it an attractive subject with which to build upon these advancements for further development of a forward genetic system in a parasitic nematode (Grant et al., 2006a).
It is hoped that in reading this chapter and the one on the biology of Strongyloides spp. (Viney and Lok, 2007), C. elegans researchers will be struck by the many similarities between these parasites and their model organism and by the ease with which standard C. elegans techniques may be adapted to Strongyloides spp. Nevertheless, some aspects of the methodology outlined here, particularly those dealing with biosafety and maintenance of infected animal hosts, may seem daunting. The C. elegans Research Community should be re-assured that once an infection is established in laboratory animals, and a straightforward set of handling protocols is in place, all aspects of this methodology are eminently practical and feasible. As the specialty of laboratory animal medicine expands, institutional vivaria are increasingly staffed by attending veterinarians who can provide assistance with technical aspects of maintaining parasite cultures in dogs and rodents. Furthermore, C. elegans researchers are frequently located in biomedical institutions that also include parasitologists. In these cases especially, productive collaborations may be formed that facilitate both the technical aspects of the research and the application of basic science.
The approaches detailed in this chapter were developed through fruitful collaborations and exchange of ideas with Gerhard Schad, Tom Nolan Ariel Junio and Meera Sundaram at Penn, Mark Viney at the University of Bristol and Warwick Grant at AgResearch, Ltd., New Zealand. The generosity of all these individuals in sharing ideas, materials and methods is gratefully acknowledged. Work in the author's laboratory was supported by grants from NIH (AI50688, AI22662 and RR02512) and the Ellison Medical Foundation (ID-IA-0037-02).
Aboobaker, A.A., and Blaxter, M.L. (2003). Use of RNA interference to investigate gene function in the human filarial nematode parasite Brugia malayi. Mol. Biochem. Parasitol. 129, 41–51. Abstract Article
Ashton, F.T., Bhopale, V.M., Holt, D., Smith, G., and Schad, G.A. (1998). Developmental switching in the parasitic nematode Strongyloides stercoralis is controlled by the ASF and ASI amphidial neurons. J. Parasitol. 84, 691–695. Abstract Article
Ashton, F.T., Zhu, X., Boston, R., Lok, J.B., and Schad, G.A. (2007). Strongyloides stercoralis: Amphidial neuron pair ASJ triggers significant resumption of development by infective larvae under host-mimicking in vitro conditions. Exp. Parasitol. 115, 92–97. Abstract Article
Bargmann, C.I., and Avery, L. (1995). Laser killing of cells in Caenorhabditis elegans. In Caenorhabditis elegans: Modern Biological Analysis of an Organism, H.F. Epstein, and D.C. Shakes, eds. (San Diego, CA: Academic Press, Inc.), pp. 225–250. Abstract
Blaxter, M.L., De Ley, P., Garey, J.R., Liu, L.X., Scheldeman, P., Vierstraete, A., Vanfleteren, J.R., Mackey, L.Y., Dorris, M., Frisse, L.M., et al. (1998). A molecular evolutionary framework for the phylum Nematoda. Nature 392, 71–75. Abstract Article
Bowman, D.D. (1995). Georgi's Parasitology for Veterinarians. In (Philadelphia: W.B. Saunders), pp. 150–245.
Fire, A., Xu, S., Montgomery, M.K., Kostas, S.A., Driver, S.E., and Mello, C.C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. Abstract Article
Ford, L., Guiliano, D.B., Oksov, Y., Debnath, A.K., Liu, J., Williams, S.A., Blaxter, M.L., and Lustigman, S. (2005). Characterization of a novel filarial serine protease inhibitor, Ov-SPI-1, from Onchocerca volvulus, with potential multifunctional roles during development of the parasite. J. Biol. Chem. 280, 40845–40856. Abstract Article
Gardner, M.P., Gems, D., and Viney, M.E. (2006). Extraordinary plasticity in aging in Strongyloides ratti implies a gene-regulatory mechanism of lifespan evolution. Aging Cell 5, 315–323. Abstract Article
Grant, W.N., Stasiuk, S., Newton-Howes, J., Ralston, M., Bisset, S.A., Heath, D.D., and Shoemaker, C.B. (2006a). Parastrongyloides trichosuri, a nematode parasite of mammals that is uniquely suited to genetic analysis. Int. J. Parasitol. 36, 453–466. Abstract Article
Grant, W.N., Skinner, S.J.M., Howes, J.N., Grant, K., Shuttle worth, G., Heath, D.D., and Shoemaker, C.B. (2006b). Heritable transgenesis of Parastrongyloides trichosuri: A nematode parasite of mammals. Int. J. Parasitol. 36, 475–483. Abstract Article
Gray, J.M., Hill, J.J., and Bargmann, C.I. (2005). A circuit for navigation in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 102, 3184–3191. Abstract Article
Gu, T., Orita, S., and Han, M. (1998). Caenorhabditis elegans SUR-5, a novel but conserved protein, negatively regulates LET-60 Ras activity during vulval induction. Mol. Cell. Biol. 18, 4556–4564. Abstract
Hashmi, S., Zhang, J., Oksov, Y., and Lustigman, S. (2004). The Caenorhabditis elegans cathepsin Z-like cysteine protease, Ce-CPZ-1, has a multifunctional role during the worms' development. J. Biol. Chem. 279, 6035–6045. Abstract Article
Hawdon, J.M., and Schad, G.A. (1991). Long term storage of hookworm infective larvae in buffered saline solution maintains larval responsiveness to host signals. J. Helminthol. Soc. Wash. 58, 140–142.
Higazi, T.B., Merriweather, A., Shu, L., Davis, R., and Unnasch, T.R. (2002). Brugia malayi: transient transfection by microinjection and particle bombardment. Exp. Parasitol. 100, 95–102. Abstract Article
Hotez, P., Molyneux, D.H., Fenwick, A., Ottesen, E.A., Sachs, S.E., and Sachs, J.D. (2006). Incorporating a rapid-impact package for neglected tropical diseases with programs for HIV/AIDS, tuberculosis, and malaria. PLoS Med. 3, 0001–0009. Abstract Article
Hussein, A.S., Kichenin, K., and Selkirk, M.E. (2002). Suppression of secreted acetylcholinesterase expression in Nippostrongylus brasiliensis by RNA interference. Mol. Biochem. Parasitol. 122, 91–94. Abstract Article
Issa, Z., Grant, W.N., Stasiuk, S., and Shoemaker, C.B. (2005). Development of methods for RNA interference in the sheep gastrointestinal parasite, Trichostrongylus colubriformis. Int. J. Parasitol. 35, 935–940. Abstract Article
Jackstadt, P., Wilm, T.P., Zahner, H., and Hobom, G. (1999). Transformation of nematodes via ballistic DNA transfer. Mol. Biochem. Parasitol. 103, 261–266. Abstract Article
Jansen, G., Thijssen, K.L., Werner, P., van der Horst, M., Hazendonk, E., and Plasterk, R.H. (1999). The complete family of genes encoding G proteins of Caenorhabditis elegans. Nat. Genet. 21, 414–419. Abstract Article
Johnson, N.M., Behm, C.A., and Trowell, S.C. (2005). Heritable and inducible gene knockdown in C. elegans using Wormgate and the ORFeome. Gene 359, 26–34. Abstract Article
Kerlin, R.L., Nolan, T.J., and Schad, G.A. (1995). Strongyloides stercoralis: histopathology of uncomplicated and hyperinfective strongyloidiasis in the Mongolian gerbil, a rodent model for human strongyloidiasis. Int. J. Parasitol. 25, 411–420. Abstract Article
Lewis, J.A., and Fleming, J.T. (1995). Basic Culture Methods. In Methods in Cell Biology, vol. 48. Caenorhabditis elegans: Modern Biological Analysis of an Organism, H.F. Epstein, and D.C. Shakes, eds. (New York: Academic Press), pp. 4–29. Abstract
Li, X., Massey, H.C., Nolan, T.J., Schad, G.A., Kraus, K., Sundaram, M., and Lok, J.B. (2006). Successful transgenesis of the parasitic nematode Strongyloides stercoralis requires endogenous non-coding control elements. Int. J. Parasitol. 36, 671–679. Abstract Article
Lok, J.B., and Massey, H.C., Jr. (2002). Transgene expression in Strongyloides stercoralis following gonadal microinjection of DNA constructs. Mol. Biochem. Parasitol. 119, 279–284. Abstract Article
Mello, C., and Fire, A. (1995). DNA transformation. Methods Cell Biol. 48, 451–482. Abstract
Newton-Howes, J., Heath, D.D., Shoemaker, C.B., and Grant, W.N. (2006). Characterisation and expression of an Hsp70 gene from Parastrongyloides trichosuri. Int. J. Parasitol. 36, 467–474. Abstract Article
Nolan, T.J., Bhopale, V.M., and Schad, G.A. (1999). Hyperinfective strongyloidiasis: Strongyloides stercoralis undergoes an autoinfective burst in neonatal gerbils. J. Parasitol. 85, 286–289. Abstract Article
Nolan, T.J., Megyeri, Z., Bhopale, V.M., and Schad, G.A. (1993). Strongyloides stercoralis: the first rodent model for uncomplicated and hyperinfective strongyloidiasis, the Mongolian gerbil (Meriones unguiculatus). J. Infect. Dis. 168, 1479–1484. Abstract
Nolan, T.J., and Schad, G.A. (1996). Tacrolimus allows autoinfective development of the parasitic nematode Strongyloides stercoralis. Transplantation 62, 1038. Abstract Article
Praitis, V., Casey, E., Collar, D., and Austin, J. (2001). Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157, 1217–1226. Abstract
Schad, G.A. (1989). Morphology and life history of Strongyloides stercoralis. In Strongyloidiasis: a Major Roundworm Infection of Man. D.I. Grove, ed. (Philadelphia: Taylor & Francis Inc.), pp. 85–104. Abstract
Schad, G.A., Aikens, L.M., and Smith, G. (1989). Strongyloides stercoralis: is there a canonical migratory route through the host? J. Parasitol. 75, 740–749. Article
Tabara, H., Grishok, A., and Mello, C.C. (1998). RNAi in C. elegans: soaking in the genome sequence. Science 282, 430–431. Abstract Article
Tavernarakis, N., Wang, S.L., Dorovkov, M., Ryazanov, A., and Driscoll, M. (2000). Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nat. Genet. 24, 180–183. Abstract Article
Timmons, L., Court, D.L., and Fire, A. (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263, 103–112. Abstract Article
Viney, M.E. (1999). Exploiting the life cycle of Strongyloides ratti. Parasitol. Today 15, 231–235. Abstract Article
Viney, M.E., Green, L.D., Brooks, J.A., and Grant, W.N. (2002). Chemical mutagenesis of the parasitic nematode Strongyloides ratti to isolate ivermectin resistant mutants. Int. J. Parasitol. 32, 1677–1682. Abstract Article
Viney, M.E., Matthews, B.E., and Walliker, D. (1992). On the biological and biochemical nature of cloned populations of Strongyloides ratti. J. Helminthol. 66, 45–52. Abstract
*Edited by Ralf J. Sommer. WormMethods editor Victor Ambros. Last revised December 21, 2006. Published February 17, 2007. This chapter should be cited as: Lok, J.B. Strongyloides stercoralis: a model for translational research on parasitic nematode biology (February 17, 2007), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.134.1, http://www.wormbook.org.
Copyright: © 2007 James B. Lok. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: jlok@vet.upenn.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.