Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
C. elegans has distinguishing characteristics that predestine this organism for the visualization of gene expression patterns using the reporter genes green fluorescent protein (GFP; Chalfie et al., 1994) and βGAL (LacZ; Fire et al., 1990). The transparency of C. elegans allows for microscopic analysis in vivo without animal dissection. In addition, the relative thinness of animals (approximately 75 μm cross-section) often alleviates the need for high-powered confocal microscopy. Finally, germline transformation techniques allow for the rapid generation of transgenic animals.
The E. coli gene LacZ, encoding β-galactosidase, was first used for single-cell gene expression analysis in C. elegans in 1990 (Harrison et al., 1990) and was the reporter gene of choice until the introduction of GFP in 1994 (Chalfie et al., 1994). The primary advantage of GFP over LacZ is the ability to visualize reporter gene expression in live animals rather than in fixed preparations. Therefore, GFP can be more easily used for a variety of purposes including (1) expression pattern analysis, (2) dissection of cis-regulatory sequences, (3) protein localization, (4) visualization of cellular anatomy (e.g., neuroanatomy), (5) cell identification, and (6) visualization of cellular and physiological processes (Chalfie et al., 1994; Hobert and Loria, 2005). For these reasons, GFP reporter transgenes have become the primary tool for gene expression analysis in C. elegans. We will focus on GFP from now on, keeping in mind that in many cases LacZ could substitute for GFP. In this chapter, we describe protocols for the generation of GFP reporter constructs and provide several technical considerations.
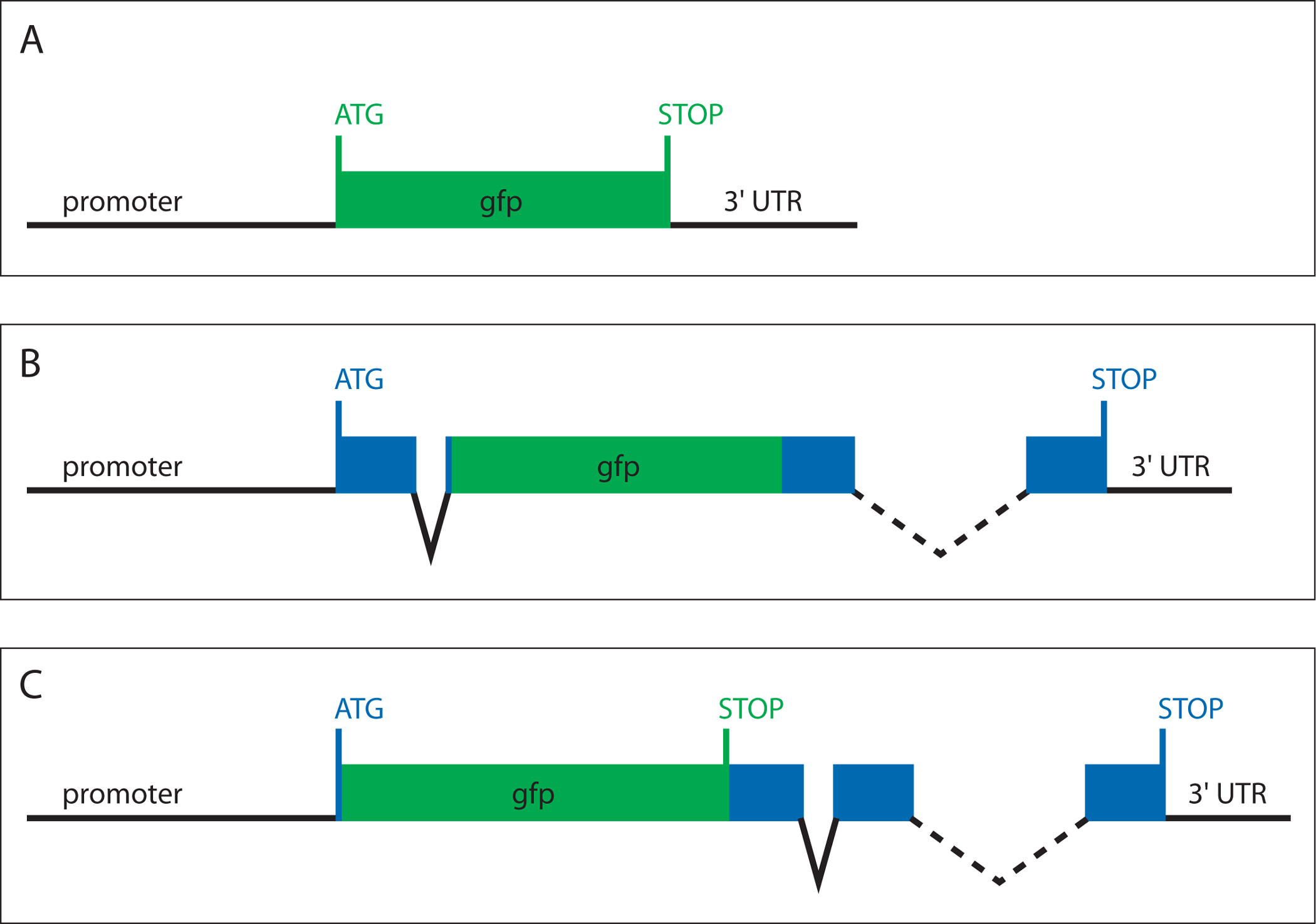
A crucial aspect of gene expression studies is choosing an adequate type of reporter for the purposes of the experiment. Each reporter differs in the amount of information it provides about the expression of a gene. The three general types of reporter gene constructs are: 1) transcriptional reporters, 2) translational reporters, and 3) "smg-1-based" transcriptional reporters (Figure 1).
Transcriptional reporters consist of a promoter fragment from a gene of interest driving GFP (Figure 1A). Typically, promoter fragments of a few kilobases immediately upstream of the start codon contain a significant portion of the cis-regulatory information necessary to provide a tentative expression pattern of the endogenous gene under study.
|
Figure 1. Architecture of reporter gene constructs. (A) Transcriptional reporter, (B) translational reporter, (C) "smg-1-based" reporter. Non-coding DNA sequences are indicated by black lines. Exon sequences are represented as blue boxes. GFP is shown in green.
Translational reporters are in-frame gene fusions between GFP and a gene of interest (Figure 1B). Ideally, a translational reporter includes the entire genomic locus of a gene (5′ upstream region, exons, introns, 3′ UTR). GFP can be inserted at any point in the open reading frame, preferably at a site that does not disrupt protein function or topology.
smg-1-based reporters are transcriptional reporters that aim to include all cis-regulatory information of a gene. GFP, including its stop codon, is inserted between the promoter and the first exon (Figure 1C). Hence, the transcription of the transgene is controlled by the cis-regulatory sequences found upstream, intronically or downstream of the gene of interest, yet only GFP is translated. However, inclusion of the GFP stop codon targets the produced mRNA for nonsense-mediated mRNA decay (Pulak and Anderson, 1993; Wilkinson et al., 1994; Mango, 2001). Therefore, these reporters need to be injected into a genetic background deficient for nonsense-mediated mRNA decay, such as smg-1 (Mango, 2001).
The C. elegans genome is very compact. Most cis-regulatory information lies within several kilobases immediately upstream of a gene. However, there is a growing number of examples for which crucial cis-regulatory sequences have been found within introns (especially genes containing large introns; Wenick and Hobert, 2004) or the 3′ UTR (Wightman et al., 1993). Rarer yet, cis-regulatory sequences have been found at unusually long distances (beyond upstream or downstream flanking genes) from the gene of interest (Conradt and Horvitz, 1999; Wenick and Hobert, 2004).
Setting aside extreme examples, the most common reporter gene constructs simply include a large portion of the 5′ intergenic sequence (transcriptional reporter). Ideally, reporters should contain as much cis-regulatory sequence, from upstream to downstream gene, as technically possible (translational and smg-1-based reporters).
Given the availability of the full C. elegans genome sequence, several groups have undertaken genome-wide gene expression projects. Three groups are systematically generating promoter::GFP fusions and documenting the resulting expression patterns. Results and additional information are available through their respective websites:
Promoterome project: http://vidal.dfci.harvard.edu/promoteromedb/
BC C. elegans Gene Expression Consortium: http://elegans.bcgsc.ca/home/ge_consortium.html
Hope Lab Expression Patten Database: http://bgypc059.leeds.ac.uk/~web/
C. elegans Promoter/Marker Database: http://www.grs.nig.ac.jp/c.elegans/promoter/index.jsp?lang=english
Transcriptional reporters can be used to rapidly establish a tentative expression pattern for a gene of interest. Fusing 5′ upstream sequences to GFP can be done in a number of ways and usually presents no technical challenge. Compared to translational and smg-1-based reporters, however, promoter fusions may not give a complete representation of the real expression pattern of a gene, both spatially and temporally.
Translational reporters can provide a faithful representation of a gene's expression pattern because additional regulatory information that may be present in introns or 3′UTRs is included in such reporter constructs. When mutants are available, translational reporters can be used in rescue experiments. Successfull rescue of the mutant phenotype lends support for the functional relevance of the resulting expression patterns. In addition, translational gene fusions can also provide information about subcellular localization and the temporal aspects of gene regulation. Translational fusions may appear less bright than transcriptional fusions due to the intrinsic instability of the protein fused to GFP. However, insertion of GFP intragenically can sometimes disrupt protein function or even lead to toxicity of the chimeric product. Finally, translational reporters that exhibit subcellular localization can make cell type identification more difficult because the shape of the cell may not be visible (especially for neurons).
smg-1-based reporters represent expression patterns as accurately as translational reporters. GFP produced by these reporters is not localized which can facilitate cell type identification. The major disadvantage of smg-1-based reporters is that they require a smg-1 mutation in the genetic background.
A large number of vector backbones and reporter gene derivatives are available from the Fire Vector Kit constructed by Andrew Fire's lab. In addition to the basic structural elements of the expression vectors (Multiple Cloning Site, artificial intron, reporter gene, 3′ UTR; see Cloning) some vectors have been engineered to contain nuclear localization, membrane targeting or mitochondrial targeting signals. The vectors, their sequence files, detailed descriptions and instructions for using these vectors are available at: http://www.addgene.org/pgvec1?f=c&cmd=showcol&colid=1 or ftp://ftp.wormbase.org/pub/wormbase/datasets/fire_vectors. Some of the most useful vectors from the Fire Vector Kit are listed in Table 1.
Table 1. Useful vector backbones: GFP/LacZ/DsRed reporter variants
| Vector | Description | Localization |
|---|---|---|
| pPD95.75 (L2463) | GFP [S65C] | None |
| pPD16.43 | LacZ; 1xNLS | Nucleus |
| pDsRed | DsRed; Clontech | - |
| pDsRed2 | DsRed2; Clontech | - |
| pPD135.83 (L4796) | GFP [S65C]; 2xSV40-NLS | Nucleolus and Nucleus |
| pPD136.15 (L4809) | GFP [S65C]; 3xSV40-NLS | Nucleolus > Nucleus |
| pPD133.48 (L4663) | CFP[Y66W, N146I, M153T, V163A]; 1xSV40-NLS | Nucleus > Cytoplasm |
| pPD133.45 (L4660) | CFP [Y66W, N146I, M153T, V163A]::lacZ; 1xSV40-NLS | Nucleus only |
| pPD132.112 (L4643) | YFP [S65G V68A S72A T203Y]; 1xSV40-NLS | Nucleus > Cytoplasm |
| pPD133.63 (L4671) | YFP [S65G V68A S72A T203Y]::lacZ; 1xSV40-NLS | Nucleus only |
| pPD122.39 (L4058) | PAT-3-GFP[S65C]; PAT-3 transmembrane domain | Plasma membrane |
| pPD133.54 (L4665) | Mt::CFP[Y66W, N146I, M153T, V163A] | Mitochondria |
| pPD133.60 (L4667) | Mt::YFP [S65G V68A S72A T203Y] | Mitochondria |
The availability of GFP variants with non-overlapping emission spectra (Cyan-FP, Yellow-FP) has opened the possibility for multi-color labeling of C. elegans cells in vivo. CFP and YFP variants can be separated using appropriate filter sets (Miller et al., 1999). Recently, triple color combinations (CFP, YFP, DsRed) have been used successfully to label separate classes of neurons (Hutter, 2003).
Of little consequence to the experimentalist, yet fascinating in its own right, is the autofluorescence exhibited by the gut granules and nucleoli of hypodermal cells in C. elegans. These phenomena are not a result of aberrant reporter transgene expression, but occur naturally in C. elegans.
In contrast to non-GFP-related autofluorescence, GFP reporters can sometimes induce non-specific fluorescence in posterior gut cells. This observed effect is thought to be an indirect consequence of the unc-54 3′ UTR, which is used in most vectors from the Fire Kit (see http://www.addgene.org/pgvec1?f=c&cmd=showcol&colid=1 or ftp://ftp.wormbase.org/pub/wormbase/datasets/fire_vectors).
Temporal gene regulation studies rely on the ability to detect gene expression fluctuations within a narrow time window. A drawback to using fluorescent proteins for such experiments is the time required for the maturation of the fluorophore.
Studies performed in E.coli have found the t0.5 for maturation to be 27 minutes for the S65T variant of GFP (used in pPD95.75 and its derivatives; Heim et al., 1995). Evidence from C. elegans experiments suggests that GFP S65T matures at a similar rate in the worm, as do CFP and YFP variants. The red fluorescent proteins, DsRed and DsRed2, mature significantly slower. However, new variants of DsRed have been generated that are brigther, mature faster (Shaner et al., 2004) and fluoresce in C. elegans (Etchberger, unpublished).
Sometimes expression from weak promoters may require a prolonged accumulation of protein before a fluorescent signal is detectable in vivo. In these cases, antibody staining can reveal expression of the fluorescent protein before a detectable fluorescent signal is achieved.
A number of standard coinjection markers are available to label reporter gene-containing extrachromosomal arrays in C. elegans. The most commonly used marker is rol-6(su1006) (Kramer et al., 1990). rol-6(su1006) is a dominant allele that can be used to label arrays in most genetic backgrounds, allowing for easy selection and maintenance of transgenic lines. Alternative injection markers that do require specific mutants background include: pha-1, dpy-20, unc-4, lin-15 (see WormMethods section: Transformation and microinjection). Notably, GFP reporters, such as ceh-22::gfp, unc-122::gfp, elt-2::gfp, and ttx-3::gfp have been used as injection markers in their own right.
Reporter transgenes may not represent the complete expression pattern for a gene, but still provide testable hypotheses about the site of gene function. It has become standard to supplement these hypotheses with cell-type specific rescue experiments, mosaic analysis and antibody staining. For early embryo studies, in situ hybridization can also be informative.
The most common approach for generating reporter genes in C. elegans is to clone/subclone DNA fragments into reporter gene vectors. A large array of vectors have been made by Andrew Fire's lab. The ‘Fire Vector Kit’ contains many vector variants used to study C. elegans gene expression (see http://www.addgene.org/pgvec1?f=c&cmd=showcol&colid=1 or ftp://ftp.wormbase.org/pub/wormbase/datasets/fire_vectors). All vectors have a backbone based on the pUC19 plasmid and contain the E.coli ampicilin resistance gene (AmpR) and origin of replication (Fire et al., 1990). They also have convenient multiple cloning sites (MCS) and provide a number of useful reporter gene variants. In addition to the Fire Vector Kit, many expression vectors are available in the C. elegans community.
Pros of using standard cloning techniques:
Creation of reusable reagents
Availability of large sets of compatible vectors
Verification of cloned DNA by sequencing
Cons of using standard cloning techniques:
Time intensive: minimum 2-3 days
Dependence on restriction sites
Size limits for DNA insert
Toxicity to bacteria in rare cases
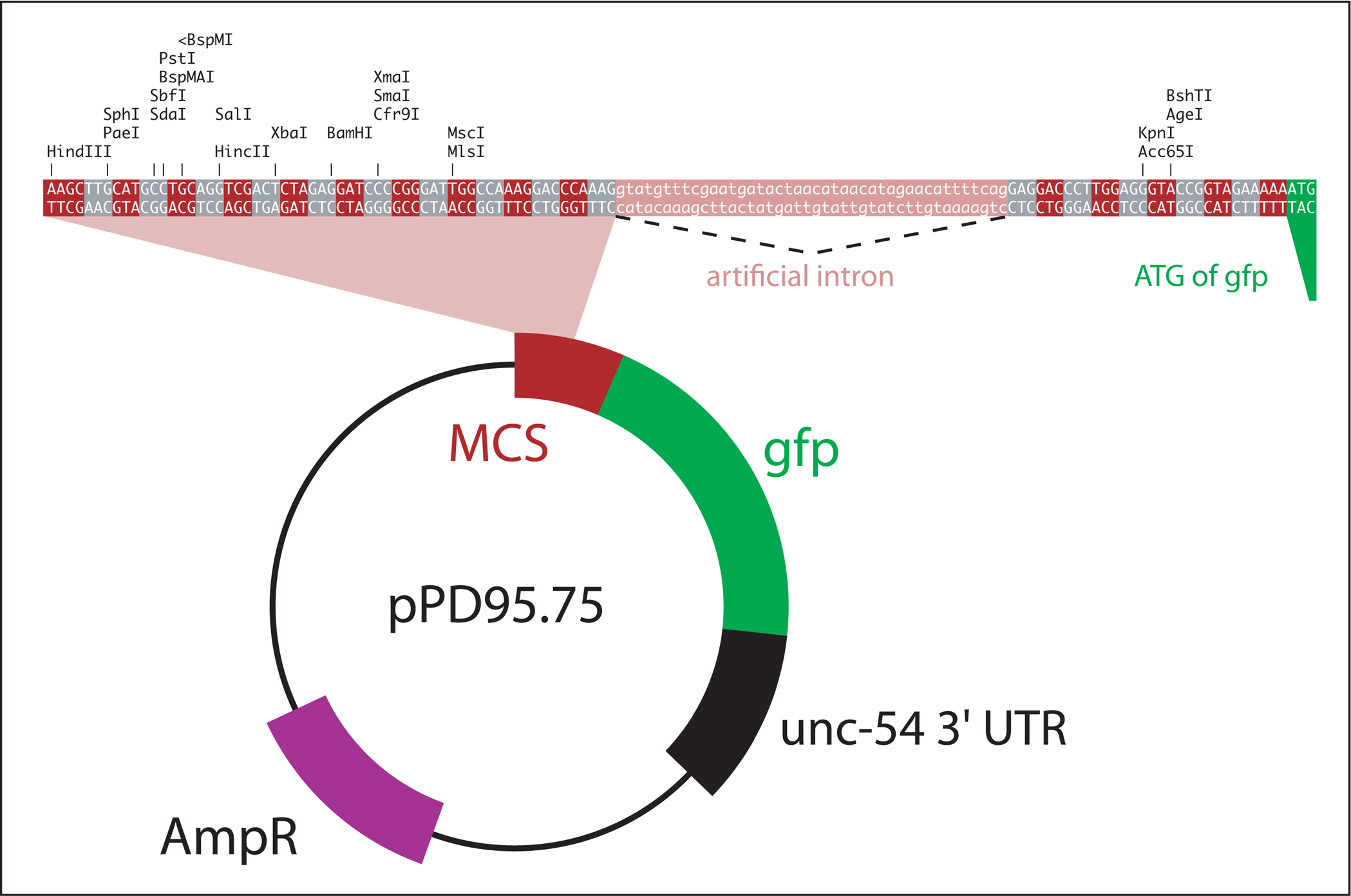
The first step in designing GFP reporter constructs is to choose an adequate vector backbone. Many vectors are available in the Fire Kit. Presented in Figure 2 is the structure of pPD95.75, a vector that serves as an archetype for most Fire vectors. In addition, a variety of specialized vectors are listed in Table 1.
The multiple cloning site of pPD95.75 stretches from HindIII to MlsI (Figure 2).
Cloning a PCR amplified promoter: adapted restriction sites can be added to facilitate directional cloning.
Generating a C-terminal translational fusion: the sequence of interest has to be cloned into the proper reading frame. The reading frame of pPD95.75 is indicated by alternating gray and red boxes in Figure 2. Vectors with shifted reading frames are also available (pPD95.77, pPD95.79).
The reporter gene in pPD95.75 is a S65C variant of GFP that includes artificial introns for enhanced expression (Fire Vector Kit, 1995). Other reporter genes are listed in Table 1.
The primary advantage of standard cloning approaches is the generation of a reusable reporter gene construct. Plasmids can be repeatedly amplified in bacteria and stored for long periods of time. In addition, constructs can be subjected to further sub-cloning steps for cis-regulatory analysis (promoter bashing) and/or functional analysis. Finally, cloned reagents can be used as a DNA source in other GFPs fusion methods.
Traditional cloning strategies involving PCR-amplified DNA fragments and GFP expression vectors are time consuming and limited by the size of DNA inserts. Current cloning/subcloning protocols require at least 2-3 days to obtain a final construct ready for use. To obtain reporter constructs more quickly and circumvent the challenge of cloning large DNA fragments, DNA sequences can be fused to GFP via the polymerase chain reaction (PCR) and injected into the C. elegans gonad in a linear form (Hobert, 2002). This approach is referred to as PCR fusion. For more details, see Figure 3 and Hobert (2002).
Pros of using the PCR fusion approach:
Rapid generation of reporter constructs
Adaptable to any vector backbone
Independent of presence/absence of restriction sites
Cons of using the PCR fusion approach:
PCR products are a depletable resource
Sequencing validation of reporter construct impractical
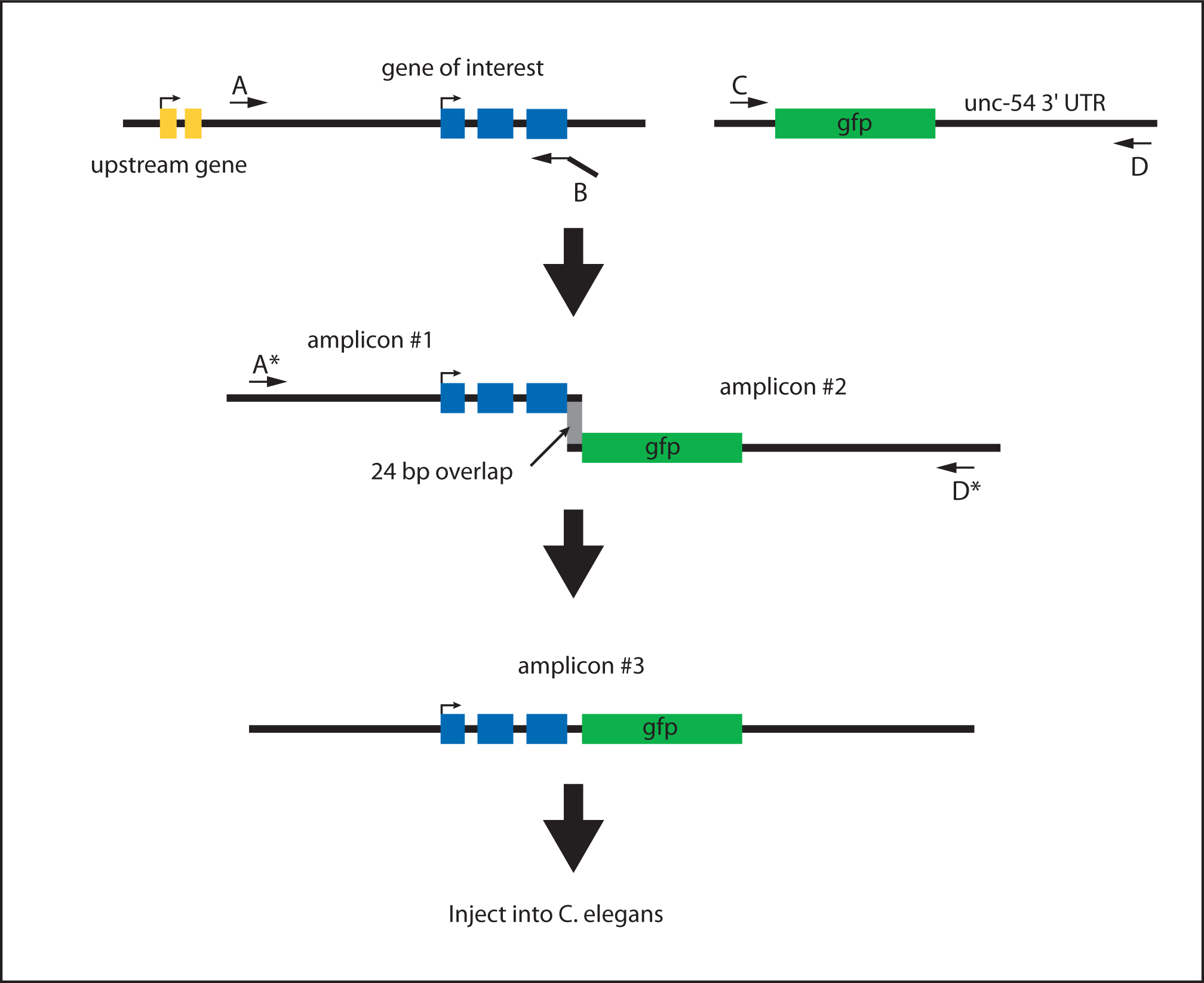
PCR fusions generate a single amplicon from two fragments that contain a small overlapping region of sequence homology (~24 bp; Figure 3). The overlap is engineered into the fragments by the primers. By this method, a sequence of interest can be fused to any reporter gene.
Many derivatives of GFP expression vectors currently exist (Fire Vector Kit). Choose a reporter gene vector that suits the needs of the experiment. pPD95.75 is a commonly used vector for PCR fusion. However, in some cases, a vector containing a localized reporter gene or 3′UTR variant may be needed (Table 1). Remember that primer design depends upon the sequence of the reporter gene vector.
The following guidelines for primer design correspond to situtations where GFP is fused to the 3′ end of a DNA fragment (e.g., promoter or genomic locus). Gene-specific primers are used to amplify the promoter and/or gene of interest (Figure 3: primers A, A*, B; A* is nested relative to A; Table 2). Likewise, vector-specific primers are used to amplify the expression vector of choice, encompassing the reporter gene and 3′ UTR (Figure 3: primers C, D, D*; D* is nested relative to D; Table 2). The primer requiring the most careful design is the 3′ gene-specific primer (Figure 3: primer B). This primer contains a linking sequence that overlaps with the reporter gene amplicon. The gene-specific primer linker should be at least 24 bp long and complementary to the MCS of the expression vector or to the coding region of the reporter gene.
Table 2. Primer sequences used with pPD95.75
| Primer A | Gene-specific 5' 'outside' primer |
| Primer A* | Gene-specific 5' 'nested' primer |
| Primer B | 5′-AGTCGACCTGCAGGCATGCAAGCT-Gene-specific sequence-3′ |
| Primer C | 5′-AGCTTGCATGCCTGCAGGTCG-3′ |
| Primer D | 5′-AAGGGCCCGTACGGCCGACTA-3′ |
| Primer D* | 5′-GGAAACAGTTATGTTTGGTATA-3′ |
|
Figure 3. Generating a C-terminal translational PCR fusion. Primers A and B amplify the genomic region (amplicon #1). Primer B adds a 24 bp overlap in frame to the GFP coding region. Primers C and D amplify the reporter gene (e.g., GFP) and 3' UTR (amplicon #2). Primers A* and D* are used to fuse amplicon #1 and amplicon #2 (gray box indicates 24 bp sequence overlap). The resulting fusion product (amplicon #3) can be directly injected into C. elegans without purification.
Steps:
PCR amplification of genomic DNA and reporter gene
Amplify DNA fragment with gene-specific primers (Primers A and B) from a genomic DNA preparation, worm lysate, cosmid or other source.
Amplify reporter gene from expression vector with vector-specific primers (for pPD95.75 Primers C and D).
Estimate DNA concentration of PCR products by agarose gel electrophoresis.
Fusion PCR
Fuse products from Step 1 (1-50 ng each per reaction) using nested primers (Primer A*; for pPD95.75 Primer D*).
Estimate DNA concentration of PCR product by agarose gel electrophoresis.
Sample preparation and injection
The PCR reaction from Step 2 can be used directly for injection if the expected size fusion product is present in sufficient quantity. If yield is low, the DNA sample can be concentrated by standard methods. Usually, reporters are injected at concentrations ranging from 10 to 50 ng/μL.
PCR fusion technology is a good supplement to other strategies for the generation of reporter gene constructs. The primary advantage to the PCR fusion approach is speed. The time required to make a PCR fusion product from start to finish is shorter than traditional cloning approaches. In addition, many reactions can be carried out in parallel. However, PCR fusion technology is not to replace all traditional cloning strategies. Most significantly, PCR fusion products are not amplifiable in bacteria. Furthermore, additive rounds of PCR reactions increase the chances of acquiring point mutations in the amplicon. This problem can be reduced by pooling independent fusion reactions.
Reporter gene constructs for specific genomic loci can be extremely long (>20kb). The manipulation of large genomic fragments containing complete or partial open reading frames as well as 5′ and 3′ intergenic regions can be difficult with current cloning or PCR approaches. To circumvent these limitations, DNA fragments with overlapping sequence homology can be coinjected and will undergo homologous recombination in vivo to produce a long linear fragment (Mello et al., 1991; Maryon et al., 1996; Tsalik et al., 2003). This approach is termed in vivo recombination. Reporter transgenes made in this manner eliminate the time required for conventional cloning techniques and bypass limitations associated with the PCR-mediated amplification of long DNA fragments.
Pros of using in vivo recombination:
Generation of large reporter transgenes
Cons of using in vivo recombination:
Validation of reporter transgene sequence difficult
Multiple control injections required
In vivo recombination circumvents size limitations associated with cloning and PCR fusion approaches. Smaller fragments with a minimum overlap of 350bp (Maryon et al., 1996), can be generated by various methods and coinjected to recombine into the full reporter transgene. All three types of reporter trangenes (transcriptional, translational and "smg-1-based") can be generated by this method. As an example of this approach, the following protocol describes the generation an N-terminally fused GFP translational reporter.
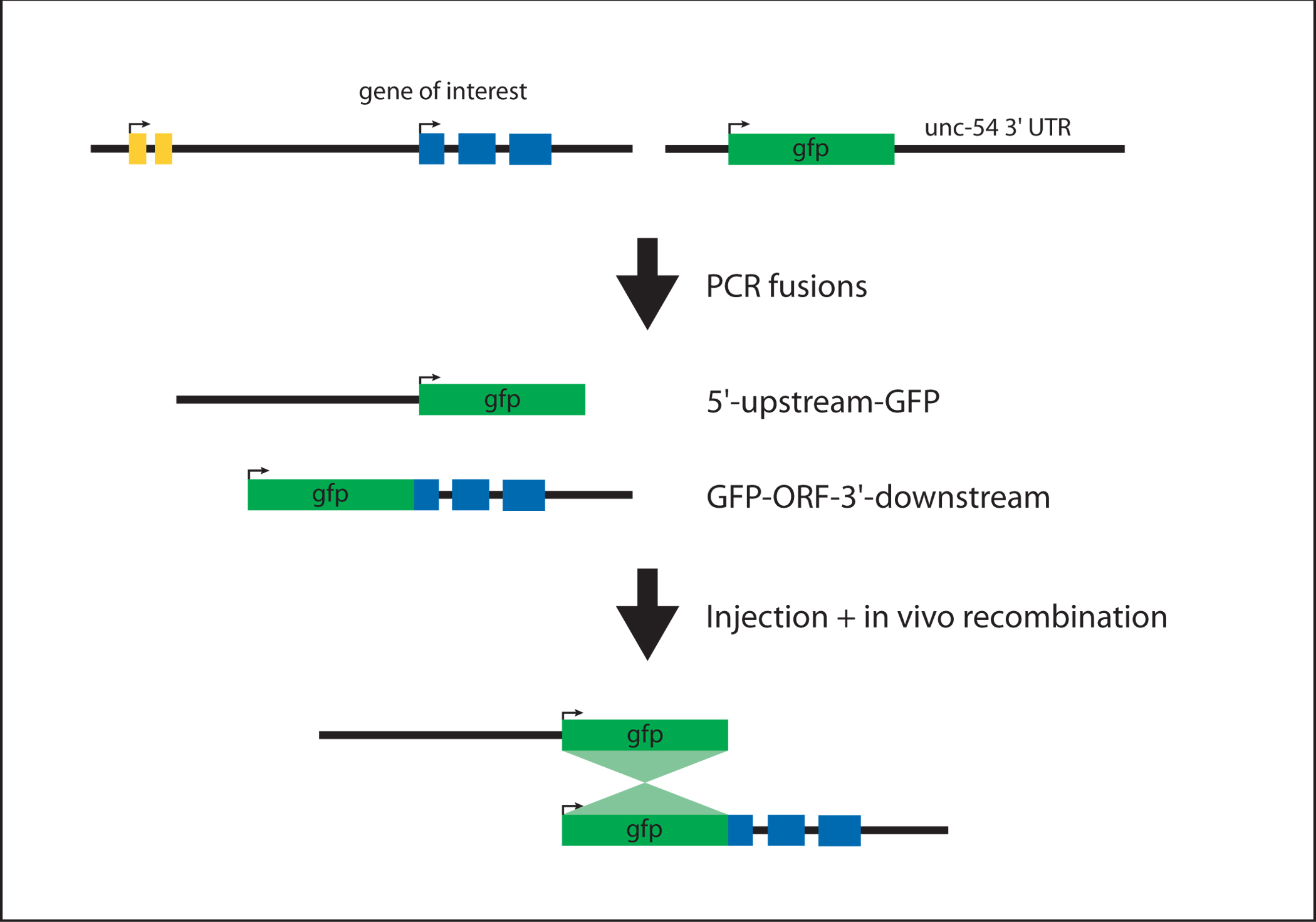
To generate a translational GFP reporter for a large gene (e.g., lin-41; pers. com. Frank Slack), the simplest approach is to generate 5′-upstream-GFP and GFP-ORF-3′-downstream fragments (by PCR fusion in this case) which will recombine to form 5′-upstream-GFP-ORF-3′-downstream (see Figure 4). 5′-upstream-GFP contains the promoter fused to the coding region of GFP. GFP-ORF-3′-downstream is a fusion of GFP to the genomic coding region and the complete 3′ UTR.
|
Figure 4. Generating a N-terminal translational reporter. The 5'-upstream-GFP and GFP-ORF-3'-downstream fragments are generated in two independent PCR fusion steps (see protocol #2). The products are injected at equal molar ratios. The sequence overlap between the two fragments (GFP coding region) allows for homologous recombination in vivo to generate 5'-upstream-GFP-ORF-3'-downstream.
Steps:
5′-upstream-GFP fragment
PCR amplify 5′-upstream to include overlapping 3′ linker to the first 24nt of the GFP coding region (GFP sequence: ATGAGTAAAGGAGAAGAACTTTTC).
PCR amplify GFP coding region (5′ primer: ATGAGTAAAGGAGAAGAACTTTTC; 3′ primer: GTCAGAGGCACGGGCGCGAGATG) from reporter gene vector (e.g., pPD95.75).
PCR fuse 5′-upstream and GFP coding region not including the STOP codon (5′ nested primer; 3′ nested GFP primer: TTTGTATAGTTCATCCATGCC).
GFP-ORF-3′-downstream fragment
PCR amplify ORF-3′-downstream to include overlapping 5′ linker to the last 24nt of the GFP coding region minus the STOP codon (GFP sequence: CATGGCATGGATGAACTATACAAA).
PCR amplify GFP coding region (5′ primer: GTTTCGAATGATACTAACATAAC; 3′ primer: TTTGTATAGTTCATCCATGCC) from reporter gene vector (e.g., pPD95.75).
PCR fuse GFP coding region and ORF-3'-downstream region (5′ nested GFP primer: ATGAGTAAAGGAGAAGAACTTTTC; 3′ nested ORF-3′-downstream primer).
Co-injection of PCR fusion products
Inject PCR fusion products (5′-upstream-GFP and GFP-ORF-3′-downstream) simultaneously at equal molar ratios.
In vivo recombination technology provides a way to bypass the difficulties involved in the amplification and cloning of large DNA fragments. However, the efficiency of homologous recombination in C. elegans is largely uncharacterized. In addition, the 5′-upstream-GFP or GFP-ORF-3′-downstream fragment may lead to erroneous expression on its own. To minimize undesirable background expression, truncated GFP sequences that maintain sufficient overlap to trigger homologous recombination could be used.
In the wake of large scale genome sequencing projects, there has been an effort to make available the large number of functionally relevant DNA sequences (e.g., ORFs, cDNAs, promoters, 3′ UTRs) in a versatile format. Gateway cloning technology http://www.invitrogen.com/content.cfm?pageid=4072 is a high-throughput method available for in vitro cloning of DNA fragments into a variety of vector backbones.
Presented here is a Gateway cloning strategy suitable for generating transcriptional promoter::gfp fusions and translational promoter::ORF::gfp fusions (Dupuy et al., 2004; Hope et al., 2004). In addition, the promoter clones generated by this method can be used within the Gateway system for a variety of applications (e.g., tissue-specific RNAi and yeast one hybrid screens).
Pros of using Gateway® technology:
Fast, efficient in vitro reaction
Compatible with large collection of vectors for other studies (Dupuy et al., 2004)
Cons of using Gateway® technology:
Decreased cloning efficiency above 2-3 kb (Marsischky and LaBaer, 2004)
Proprietary cloning system
Multistep procedure
The Gateway system takes advantage of the site-specific recombination properties of bacteriophage lambda (Landy, 1989; Hartley et al., 2000). Bacteriophage lambda integrase proteins recognize compatible recombination sites (att sites), found in the cloning vector and flanking the DNA sequence of interest, to carry out the recombination event.
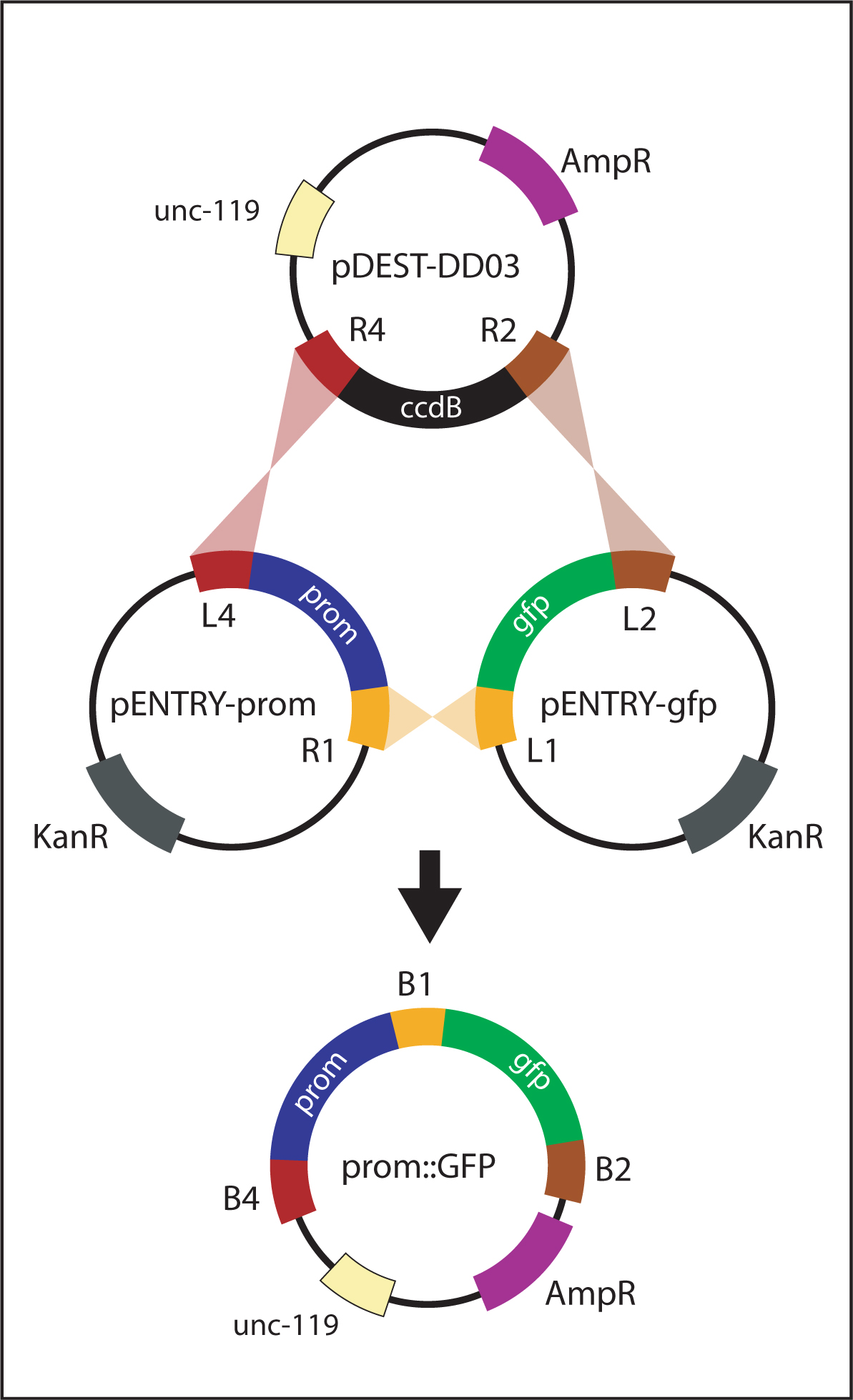
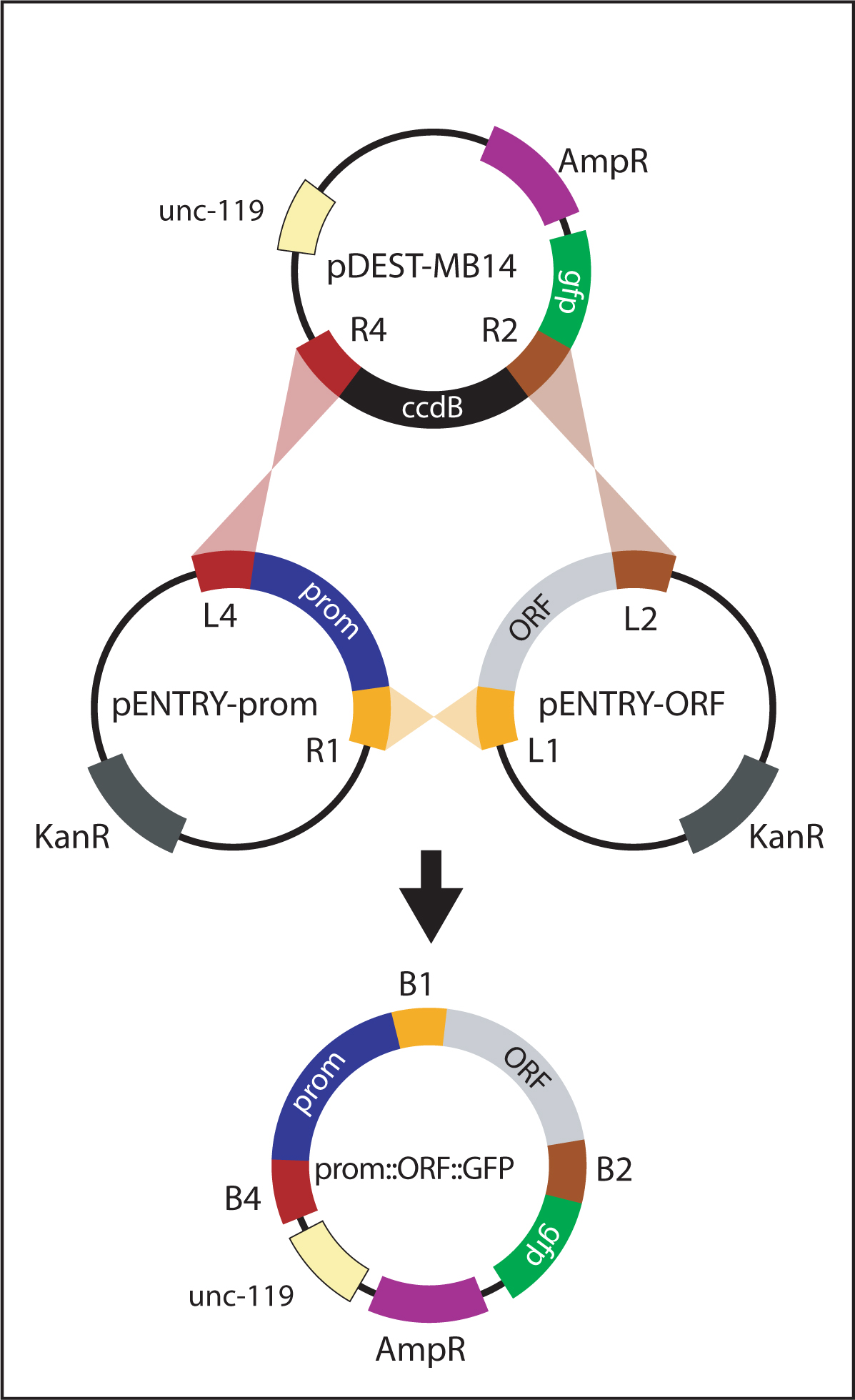
For generating promoter::gfp and promoter::ORF::gfp reporters, a promoter is initially amplified with primers containing flanking att sites (attB4 and attB1R). The PCR product is recombined with pDONR-P4-P1R to generate pENTRY-prom (Figure 5). Finally, pENTRY-prom is recombined with the appropriate vectors to generate the intended product:
| promoter::gfp reporter: recombine pENTRY-prom with pENTRY-gfp and pDEST-DD03 (Figure 6). |
| promoter::ORF::gfp reporters: recombine pENTRY-prom with pENTRY-ORF and pDEST-MB14 (Figure 7). |
|
Figure 5. Generating pENTRY-prom. The attB4-prom-attB1R fragment is amplified with appropriate primers containing attB4 and attB1R sequences. attB4 and attB1R recombine with attP4 and atP1R of pDONR-P4-P1R, respectively, to generate pENTRY-prom in which the promoter is flanked by attL4 and attR1 sites.
|
Figure 6. Generating promoter::gfp. The promoter::gfp vector is generated by a multi-site LR reaction between pENTRY-prom, pENTRY-gfp and pDEST-DD03. attL sites recombine with corresponding attR sites. The resulting plasmid, promoter::gfp, contains the promoter and GFP flanked by attB sites.
|
Figure 7. Generating promoter::ORF::gfp. The promoter::ORF::gfp vector is generated by a multi-site LR reaction between pENTRY-prom, pENTRY-ORF and pDEST-MB14. In this reaction, attL sites recombine with corresponding attR sites. The resulting plasmid, promoter::ORF::gfp, contains the promoter, ORF and GFP in frame with each other. pENTRY-ORF vectors are specifically designed to recombine in frame with GFP from pDEST-MB14.
Steps:
Generation of attB4-prom-attB1R
Design primers to amplify a promoter of interest. The primers must contain attB4 (5′ primer) and attB1R (3′ primer) sequences in addition to the promoter specific sequences.
5′ primer: (attB4 site)
5′-GGGGCAACTTTGTATAGAAAAGTTG---promoter-specific sequence-3′
3′ primer: (attB1R site)
5′-GGGGACTGCTTTTTTGTACAAACTTGTC---promoter-specific sequence-3′
Amplify attB4-prom-attB1R from an appropriate source (genomic DNA preparation, worm lysate, plasmid, cosmid, etc.).
Generation of pENTRY-prom
Recombine attB4-prom-attB1R with pDONR-P4-P1R (Invitrogen, cat. num. 12537023) to generate an Entry Vector (pENTRY-prom) in which the promoter is flanked by attL4 and attR1 sites.
Generation of promoter::gfp and promoter::ORF::gfp reporter
In the study by Dupuy et al., the destination vectors (pDEST-DD03 & pDEST-MB14) contain the unc-119 gene which is used in ballistic transformation protocols (Praitis, 2001). pENTRY-GFP, pENTRY-ORF, pDEST-DD03 and pDEST-MB14 can be obtained from Marc Vidal (marc_vidal@dfci.harvard.edu).
Promoterome database: http://vidal.dfci.harvard.edu/promoteromedb/
Invitrogen: http://www.invitrogen.com
Gateway cloning technology: http://www.invitrogen.com/content.cfm?pageid=4072
The analysis of gene expression from large genomic loci requires the construction of large reporter transgenes. Traditional cloning strategies rely on available restriction sites for the insertion of a reporter gene within a gene of interest. The multitude of restriction sites within large DNA fragments makes this a difficult task. One method to bypass the need for restriction sites is yeast homologous recombination (Hawkins et al., 2003). This approach exploits the naturally occuring homologous recombination in S. cerevisiae and can be used to generate precise insertions of a reporter gene into virtually any target sequence.
Pros of using yeast homologous recombination:
Generation of large (> 18kb) reporter gene fusions
No requirement for restriction sites
Limited use of PCR amplification
Cons of using yeast homologous recombination:
Requirement for yeast culturing skills
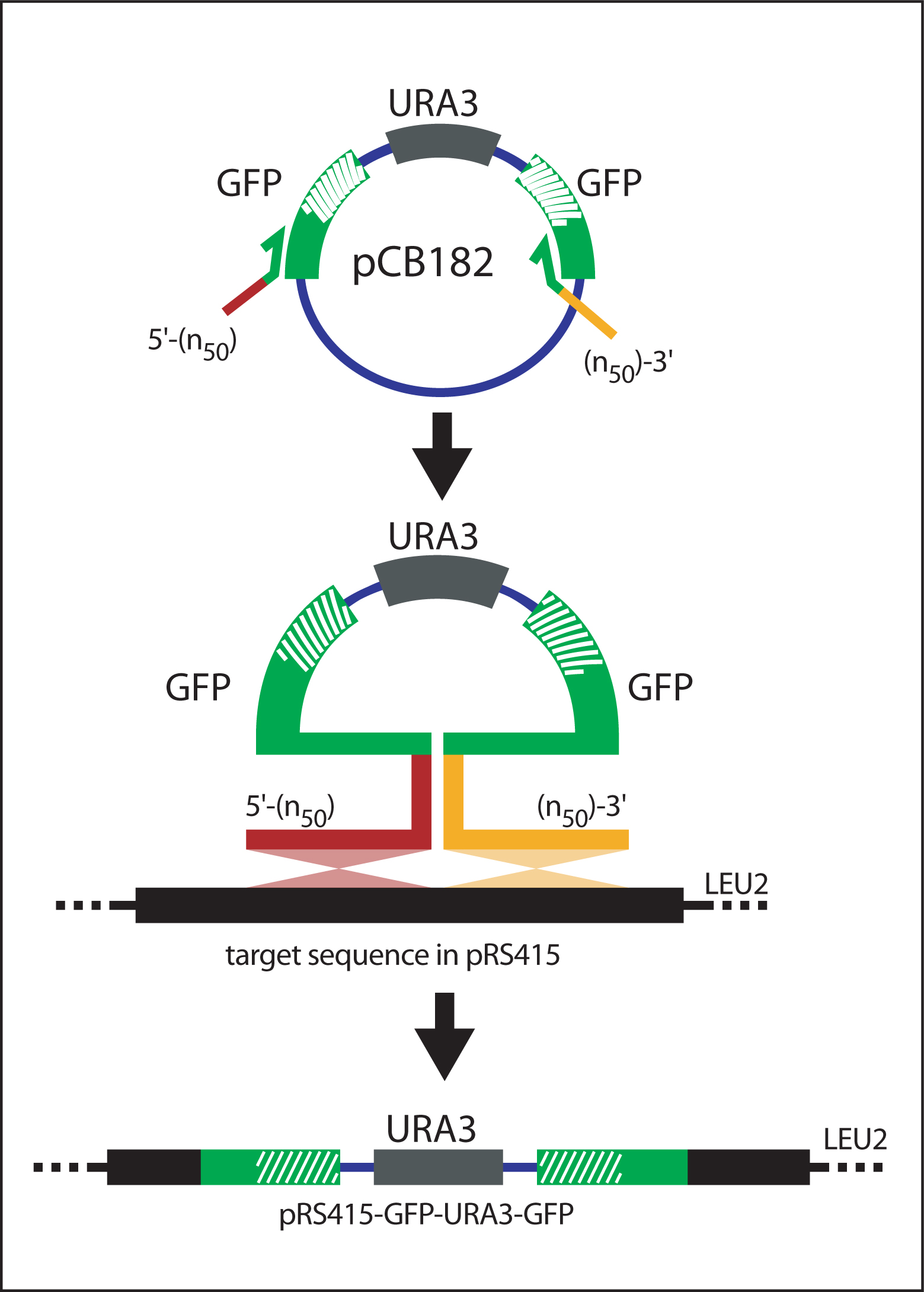
Yeast homologous recombination utilizes the site-specific recombination properties of S. cerevisiae. An initial PCR reaction amplifies a modified GFP reporter cassette (GFP-URA3-GFP) with primers containing at least 50 base pairs of sequence homologous to the insertion site within the gene of interest ("target sequence") (see Figure 8). The cassette is recombined into the target sequence that is carried by a bacterial shuttle vector (pRS415). Subsequent selection steps generate the final GFP fusion construct (see Figure 9).
Reagents:
Target sequence cloned into the yeast-bacterial shuttle vector (pRS415)
Primers to amplify (n50)-GFP-URA3-GFP-(n50) cassette (see Step 2)
Vector pCB182: contains two tandem repeats of GFP flanking URA3
Vector pRS415: yeast-bacterial shuttle vector
Yeast strain CBS17: MATα his3δ200 leu2δ1 ura3-52 mal hap2
Yeast strain CBY668: MATα ade2-101 leu2-3,112 ura3-52 cyh2 kar1-1
Steps:
Generation of (n50)-GFP-URA3-GFP-(n50) cassette
Design primers to amplify GFP-URA3-GFP. The primers must contain at least 50bp overhangs homologous to the target site and the core sequences (complementary to GFP) as follows:
5′ primer: 5′-(n50)-ATGAGTAAAGGAGAAGAACTTTTCAC-3′
3′ primer: 5′-(n50)-GAATTCTTTGTATAGTTCATCCATGC-3′
Amplify (n50)-GFP-URA3-GFP-(n50) from pCB182 (Figure 8).
|
Figure 8. Recombination of GFP-URA3-GFP into target sequence. The (n50)-GFP-URA3-GFP-(n50) cassette is amplified from pCB182 and transformed into yeast carrying the bacterial shuttle vector pRS415 (containing the target sequence). The gene specific extensions (n50) flanking GFP-URA3-GFP direct homologous recombination between (n50)-GFP-URA3-GFP-(n50) and pRS415. Selection for vector recombinants is achieved by plating the cells in the absence of uracil and leucine. Adapted from Hawkins et al. (2003).
Recombination of GFP-URA3-GFP into target sequence
Transform (n50)-GFP-URA3-GFP-(n50) into CBS17 (carrying target sequence-containing pRS415). Select on solid media lacking uracil (selection for GFP-URA3-GFP) and leucine (selection for target sequence-containing pRS415; Figure 9).
Selection of strains carrying a homogeneous population of GFP-URA3-GFP containing plasmids
The GFP-URA3-GFP cassette will recombine specifically into the target plasmid or unspecifically into the yeast genome. In addition, since yeast strains carry multiple copies of CEN-based plasmids (e.g., pRS415), not all of the plasmids need to contain the GFP-URA3-GFP insert for growth without uracil (Figure 9).
|
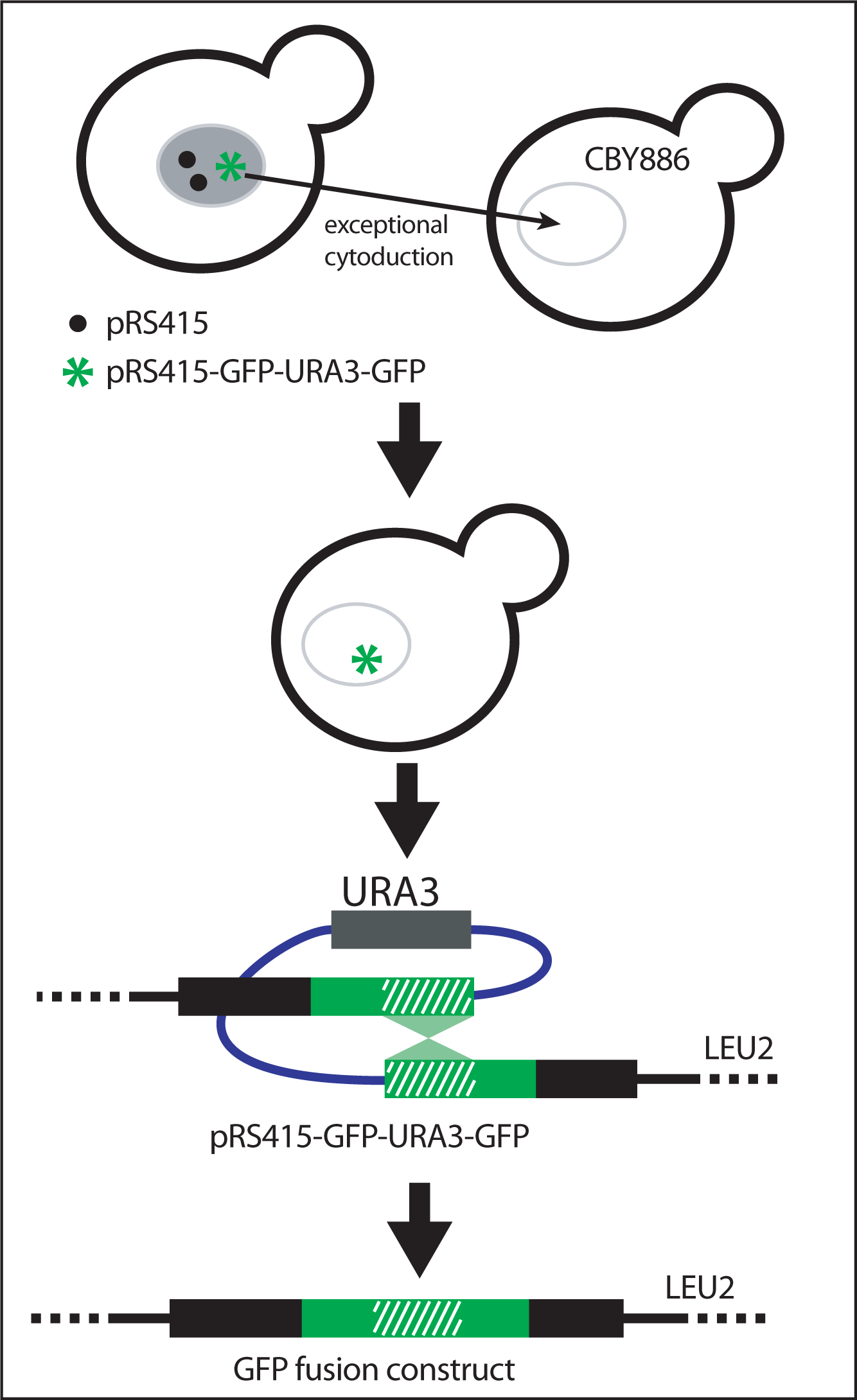
Figure 9: Isolation of GFP fusion construct. To obtain yeast carrying only the pRS415-GFP-URA3-GFP recombinant vector, plasmids are transfered by cytoduction into the karyogamy-deficient kar1-1 strain (CBY886). This step eliminates the presence of chromosomal insertions of GFP-URA3-GFP or unrecombined pRS415. Selection for the excision of URA3 and generation of the final GFP fusion product occurs when yeast are grown on on solid medium containing 5-fluoroorotic acid and lacking leucine. Adapted from Hawkins et al. (2003).
To obtain a yeast strain containing only plasmids carrying GFP-URA3-GFP and no chromosomal insertions, replica-print yeast between different solid media. Transfer individual plasmids into karyogamy-deficient recipient yeast strains (kar1-1) by exceptional cytoduction (Dutcher, 1981). The kar1-1 mutation limits the exchange of chromosomal DNA between cells but does not affect exchange of plasmids or extranuclear material. As a result, haploid cells retain their original nuclear genotype but receive plasmid from the mating partner
Mate strains carrying GFP-URA3-GFP en masse with CBY668 spread out as a lawn on rich medium. Select kar1-1 recipients on solid medium containing cycloheximide but lacking leucine. Cycloheximide resistance confered by cyh2 to CBY668 is recessive. Therefore only haploid cells will grow (not diploids and not the original transformants). Only yeast having received the plasmid grow without leucine and only yeast in which the GFP-URA3-GFP cassette was integrated into the plasmid grow without uracil.
Generation of GFP fusion construct
To select for the final GFP fusion construct, grow yeast on solid medium containing 5-fluoroorotic acid and lacking leucine (Figure 9). 5-fluoroorotic acid selects against cells containing the URA3 gene.
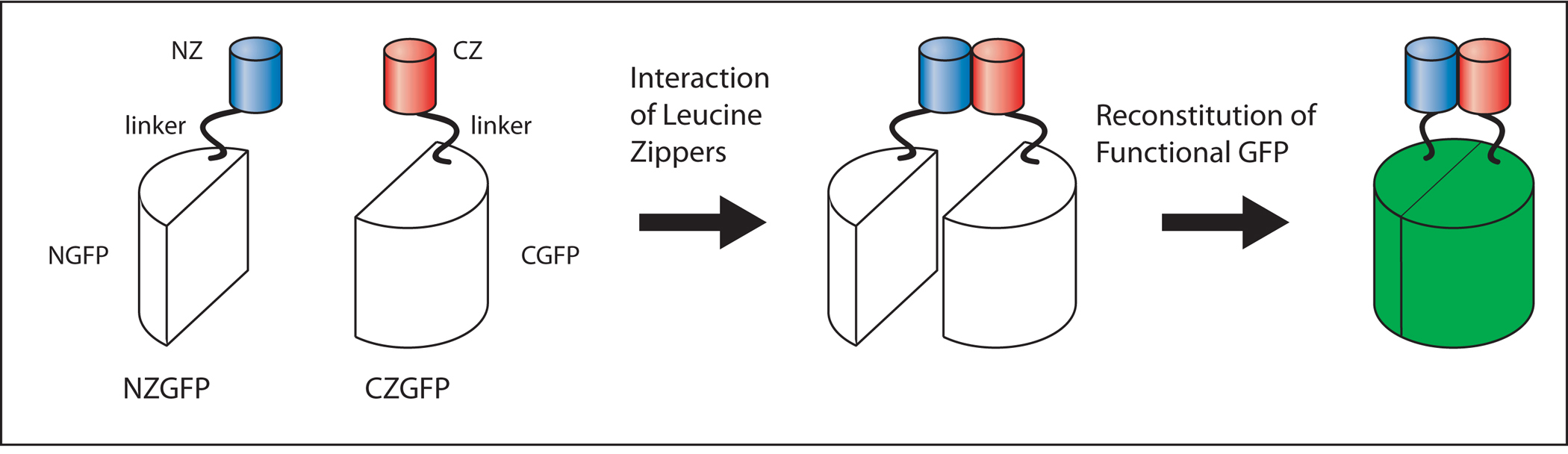
Conventional transcriptional and translational reporters rarely direct expression in a single cell type. Moreover, cell-specific cis-regulatory sequences are not available for all 959 somatic cells of C. elegans. Advances in the molecular understanding of GFP have led to the development of reconstituted GFP expression systems. Partial GFP peptides (N-GFP: residues 1-157 and C-GFP: residues 158-238) co-expressed in bacteria can form a stable fluorescent product. In eukaryotic cells, GFP fluorescence occurs when N-GFP and C-GFP are fused to short peptides (e.g., leucine zippers) that naturally interact in vivo (Ghosh et al., 2000; Figure 10; Figure 11).
|
Figure 10. NZGFP-CZGFP reconstitution. The interaction of the leucine zipper motifs (NZ, CZ) leads to the dimerization of NZGFP and CZGFP monomers. Dimerization results in the reconstitution of functional GFP (green). Adapted from Zhang et al. (2004).
|
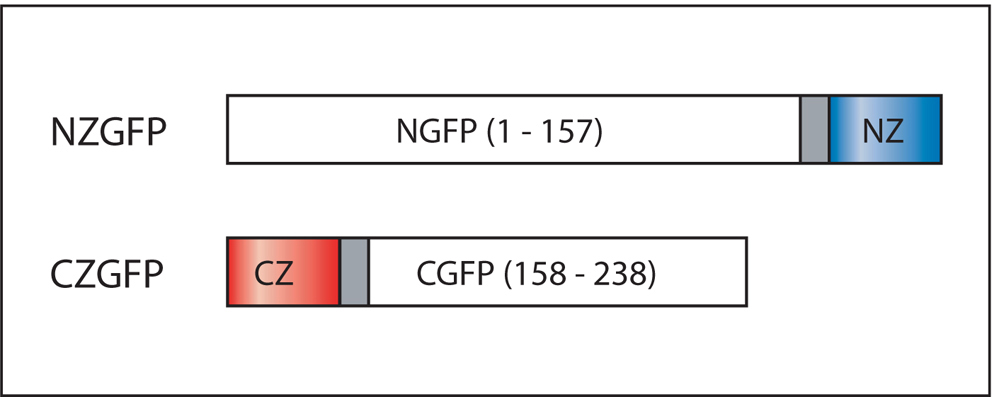
Figure 11. Domain structure of CZGFP and NZGFP. NGFP(1-157) contains a C-terminal peptide linker to the NZ leucine zipper motif. Similarly, CGFP(158-238) has a N-terminal peptide linker to the CZ leucine zipper motif. Adapted from Zhang et al. (2004).
To apply this approach to C. elegans, two expression vectors have been developed that contain GFP polypeptides, N-GFP and C-GFP, fused via a small linker to sequences that can form an antiparallel leucine zipper (NZ and CZ; Zhang et al., 2004). The vectors, refered to as NZGFP and CZGFP, are suited for the cloning of cis-regulatory DNA fragments. By combining cis-regulatory sequences yielding overlapping expression patterns, one can engineer worm strains that label specific subsets of cells.
Reagents (Chalfie, pers. comm.)
| Vector | Reporter gene | Comments |
|---|---|---|
| TU#710 | NZGFP in pPD95.77 | GFP portion in NZGFP contains the introns from the original pPD95.77 |
| TU#711 | CZGFP in pPD95.77 | GFP portion in CZGFP contains the intron from the original pPD95.77 |
| TU#712 | NZYFP version of TU#710 | Missense mutations: S65G, V68L, and S72A |
| TU#713 | CZYFP version of TU#711 | Missense mutations: T203Y |
| TU#714 | NZCFP version of TU#7102 | Missense mutations: Y66W, N146I, and M153T |
| TU#715 | CZCFP version of TU#711 | Missense mutations: V163A |
Chalfie, M., Y. Tu, et al. (1994). Green fluorescent protein as a marker for gene expression. Science 263, 802–805. Abstract
Conradt, B., and Horvitz, H.R. (1999). The TRA-1A sex determination protein of C. elegans regulates sexually dimorphic cell deaths by repressing the egl-1 cell death activator gene. Cell 98, 317–327. Abstract Article
Dupuy, D., Li, Q.R., et al. (2004). A first version of the Caenorhabditis elegans promoterome. Genome Res. 14, 2169–2175. Abstract Article
Dutcher, S.K. (1981). Internuclear transfer of genetic information in kar1-1/KAR1 heterokaryons in Saccharomyces cerevisiae. Mol. Cell Biol. 1, 245–253. Abstract
Fire, A., Harrison, S.W., et al. (1990). A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene 93, 189–198. Abstract Article
Ghosh, I., Hamilton, A.D., et al. (2000). Antiparallel leucine zipper-directed protein reassembly: application to the green fluorescent protein. J. Am. Chem. Soc. 122, 5658–5659. Article
Hartley, J.L., Temple, G.F., et al. (2000). DNA cloning using in vitro site-specific recombination. Genome Res. 10, 1788–1795. Abstract Article
Hawkins, N.C., Garriga, G., et al. (2003). Creating precise GFP fusions in plasmids using yeast homologous recombination. Biotechniques 34, 74–78, 80. Abstract
Heim, R., Cubitt, A.B., et al. (1995). Improved green fluorescence. Nature 373, 663–664. Abstract Article
Hobert, O. (2002). PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. Biotechniques 32, 728–730.Abstract
Hobert, O., and Loria, P.M. (2005). Uses of GFP in C. elegans. In: Green Fluorescent Protein: Properties, Applications and Protocols. Second edition. Chalfie and Kain, eds. (Wiley).
Hope, I.A., Stevens, J. et al. (2004). Feasibility of genome-scale construction of promoter::reporter gene fusions for expression in Caenorhabditis elegans using a multisite gateway recombination system. Genome Res. 14, 2070–2075. Abstract Article
Hutter, H. (2003). Extracellular cues and pioneers act together to guide axons in the ventral cord of C. elegans. Development 130, 5307–5318. Abstract Article
Kramer, J.M., French, R.P., et al. (1990). The Caenorhabditis elegans rol-6 gene, which interacts with the sqt-1 collagen gene to determine organismal morphology, encodes a collagen. Mol. Cell Biol. 10, 2081–2089. Abstract
Landy, A. (1989). Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu. Rev. Biochem. 58, 913–949. Abstract Article
Mango, S.E. (2001). Stop making nonsense: the C. elegans smg genes. Trends Genet. 17, 646–653. Abstract Article
Marsischky, G., and LaBaer J. (2004). Many paths to many clones: a comparative look at high-throughput cloning methods. Genome Res. 14, 2020–2028. Abstract Article
Maryon, E.B., Coronado, R., et al. (1996). unc-68 encodes a ryanodine receptor involved in regulating C. elegans body-wall muscle contraction. J. Cell Biol. 134, 885–893. Abstract Article
Mello, C.C., Kramer, J.M., et al. (1991). Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970. Abstract
Miller, D.M., III, Desai, N.S. et al. (1999). Two-color GFP expression system for C. elegans. Biotechniques 26, 914–918, 920–921. Abstract
Pulak, R., and Anderson P. (1993). mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 7, 1885–1897. Abstract
Shaner, N.C., Campbell, R.E. et al. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572. Abstract Article
Tsalik, E.L., Niacaris, T., Wenick, A.S., Pau, K., Avery, L., and Hobert, O. (2003). LIM homeobox gene-dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Dev. Biol. 263, 81–102. Abstract Article
Wenick, A.S., and Hobert, O. (2004). Genomic cis-regulatory architecture and trans-acting regulators of a single interneuron-specific gene battery in C. elegans. Dev. Cell 6, 757–770. Abstract Article
Wightman, B., Ha, I., et al. (1993). Post transcriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862. Abstract Article
*Edited by Victor Ambros. Last revised April 18, 2005. Published April 5, 2006. This chapter should be cited as: Boulin, T. et al. Reporter gene fusions (April 5, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.106.1, http://www.wormbook.org.
Copyright: © 2006 Thomas Boulin et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§Both of these authors contributed equally
†To whom correspondence should be addressed. E-mail: or38@columbia.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.