Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergAbstract
This chapter reviews analytical tools currently in use for protein classification, and gives an overview of the C. elegans proteome. Computational analysis of proteins relies heavily on hidden Markov models of protein families. Proteins can also be classified by predicted secondary or tertiary structures, hydrophobic profiles, compositional biases, or size ranges. Strictly orthologous protein families remain difficult to identify, except by skilled human labor. The InterPro and NCBI KOG classifications encompass 79% of C. elegans protein-coding genes; in both classifications, a small number of protein families account for a disproportionately large number of genes. C. elegans protein-coding genes include at least ~12,000 orthologs of C. briggsae genes, and at least ~4,400 orthologs of non-nematode eukaryotic genes. Some metazoan proteins conserved in other nematodes are absent from C. elegans. Conversely, 9% of C. elegans protein-coding genes are conserved among all metazoa or eukaryotes, yet have no known functions.
Full genome sequences make it possible, for the first time, to completely list an organism's gene products. C. elegans has ~19,800 protein-coding genes, of which ~3,400 have mutant alleles and ~2,400 others have obvious phenotypes in mass RNAi screens: this leaves ~70% of genes functionally unaccounted for. Some of these unannotated genes are clearly ancient (i.e., they encode proteins conserved in metazoa or eukaryotes) and must have critical functions, even though classical biochemistry and genetics gave no indication of them before genomics (Tatusov et al., 1997 and 2003). At least ~12,000 genes are conserved between C. elegans and C. briggsae; although most of them have no known RNAi phenotypes in culture, they must be required in some way for life in the wild (Stein et al., 2003). To make sense of these thousands of genes, it is necessary (though not sufficient) to classify their protein products en masse. Genome-wide protein classification is based on the computational analysis of primary protein sequences, which in turn is based on theories of protein structure and evolution. This chapter briefly reviews protein evolution, describes current analytical tools, and gives an overview of the C. elegans proteome. Analyses shown here are based on the WS130 archival release of WormBase.
Proteins are often classified as "homologous", "similar", or "having shared function." These three ideas are related, but are neither identical nor entirely obvious.
Similarity is the degree to which two traits correspond to one another in some way; homology is the property of two traits in different organisms being derived from a common trait in a shared ancestor (De Beer, 1997; Fitch, 2000; Ridley, 2003). Similarity can be directly observed by comparing modern proteins. Homology cannot: it can only be indirectly discerned through similarity, which requires that we have some computational model for distinguishing random from nonrandom similarity (Durbin et al., 1999). Similarity can, in principle, also arise from convergent evolution instead of divergent homology. In protein sequence analysis, it is possible to distinguish convergence from divergence by computing a phylogenetic tree, estimating the sequences of common ancestors on the tree, and checking these ancestors for increased similarity with increasing age (Fitch, 1970). Alternatively, one can detect convergence by checking proteins for dissimilarities (such as tertiary structure) that change very slowly with time (Galperin et al., 1998).
Homology can arise in three ways. To distinguish two of them, Fitch (1970) proposed the terms "orthology" and "paralogy": orthologous genes are those whose last common ancestor split into two gene lineages through speciation, while paralogous genes are those which split through intragenomic duplication within a single species. In the former case, it is likely that orthologs will go on performing similar biological roles; in the latter, paralogs have long been known to allow a second gene copy to acquire a new role by functional divergence (Fay and Wu, 2003; Taylor and Raes, 2004). Later, Gray and Fitch (1983) coined the term "xenology" to denote cases where homology arose through horizontal gene transfer (Fitch, 2000). Such transfers rarely occur into metazoa (Kurland et al., 2003), but do appear to have occurred from rhizobia to the plant-parasitic nematode Meloidogyne, and thus might have occurred from microbes to Caenorhabditis as well (Scholl et al., 2003).
Some ambiguities remain. For instance, if a single gene exists in C. elegans with several homologs in humans, and those homologs diversified after the nematode-chordate divergence, are all human homologs considered orthologs of the worm gene? One proposed solution is to call all of the genes in question inparalogs or co-orthologs (Sonnhammer and Koonin, 2002). Moreover, all of these terms treat genes as unbroken blocks of biological information. But proteins often have multiple domains which undergo intragenic duplication and rearrangement (Soding and Lupas, 2003). It is therefore possible for part of a protein to have homologies that the entire protein does not share; in SwissProt, the 50 most widely distributed protein domains can be found in 16 to 141 protein families apiece (Enright et al., 2002).
Protein functions are often assumed to be not merely similar, but unchanged, between protein orthologs, and somewhat unchanged even between paralogs. For several C. elegans proteins, this generalization has been experimentally supported (Aspöck et al., 2003; Duerr et al., 1999; Haun et al., 1998; Lee et al., 1994; Lee et al., 2001; Levitan et al., 1996; Solari et al., 2005; Westmoreland et al., 2001; Zhang et al., 1999). However, in some protein families, biochemical functions change more quickly than sequences do (Gerlt and Babbitt, 2001). Conversely, divergent protein homologs can retain common function though sharing only a few key residues (Meng et al., 2004). Furthermore, instances exist of a single biochemical function being independently generated in two or more distinct protein families through convergent evolution (Galperin et al., 1998; Morett et al., 2003). Similarity is a useful source of testable hypotheses about protein functions, but it is not a substitute for experimentally testing them.
Protein sequences are opaque to the human eye; computational analysis is required for biologists to make sense of them or sort them into groups. The first question a biologist generally asks about a new protein of interest is what known proteins are most similar to it. This problem was tamed by BLAST, which allows fast heuristic searches of large protein databases with sound statistical scores for hits (Korf et al., 2003).
While useful, BLAST searching is somewhat limited. A typical BLAST output is a jumble of pairwise alignments, often in a long list, giving only a rough sense of what the common areas of similarity are. For more clarity, one needs a coherent multiple alignment of the protein to any well-defined protein sets which it resembles. This was first addressed by scanning individual sequences with matrices of aligned protein sequences ("profiles": Gribskov et al., 1990). Later, hidden Markov models (HMMs) proved to enable sensitive and mathematically rigorous searches with aligned families (Durbin et al., 1999). This led to the development of the HMMER search software (Eddy, 2005) and its use to construct the PFAM protein family database (Bateman et al., 2004). Similar databases were developed independently (e.g., PRINTS, PROSITE, ProDom, SMART, and TIGRFAMs; Ouzounis et al., 2003); with PFAM, all of these were amalgamated into InterPro (Mulder et al., 2005). Meanwhile, BLAST was extended to accept sequence profiles as queries, allowing BLAST searches for conserved protein domains (Altschul et al., 1997; Marchler-Bauer et al., 2005).
Both BLAST and other similarity searches rely on comparing two or more primary sequences to each other and searching for statistically significant matches. However, three-dimensional protein structures can be plainly similar even where primary sequences have diverged unrecognizably, making structural similarity a powerful classification method (Huyen et al., 2004; Grant et al., 2004; Siew and Fischer, 2004). For C. elegans, only a few protein structures have been determined; most must be inferred computationally, with limited reliability (Moult et al., 2003). This situation is expected to improve through structural genomics (Chance et al., 2004; Luan et al., 2004).
Although tertiary structures are hard to predict, useful algorithms exist for detecting secondary structures. HMMs can predict signal sequences and transmembrane α-helices (Krogh et al., 2001; Nielsen and Krogh, 1998) and these predictions can be integrated for greater accuracy (Käll et al., 2004). Other programs can scan a protein sequence for potential coiled-coil regions (Lupas, 1996) or low complexity regions likely to form nonglobular domains (Promponas et al., 2000; Wan et al., 2003). In many cases, such simple features actually can suggest function. Asparagine/glutamine-rich regions may enable epigenetic regulation (Michelitsch and Weissman, 2000; Si et al., 2003). Protein regions with low sequence complexity or disordered secondary structure participate in transcriptional and translational regulation, signal transduction, and quarternary structure assembly (Dyson and Wright, 2005; Karlin et al., 2002; Liu et al., 2002). Proteins with seven predicted transmembrane sequences are often G-protein coupled receptors (Pierce et al., 2002). Coiled-coil motifs, though seemingly generic, are overrepresented in proteins required for meiosis (Colaiacovo et al., 2002). Other sequence motifs that determine subcellular localization (e.g., nuclear localization signals) have been difficult to predict with the reliability needed for genome-wide analysis; however, recent work suggests that such predictions may be feasible (Nair and Rost, 2004; Park and Kanehisa, 2003; Scott et al., 2004).
Protein size is so simple a classification that it is often overlooked. C. elegans proteins have an unsurprising median size (343 residues), but a wide size range (16-18,562 residues). Some proteins, such as cytoskeletal or extracellular matrix components, must be over 1000 residues long to do their jobs at all (e.g., dystrophin and titin; Hutter et al., 2000). Others are so small (30-80 residues) that they can barely support stable tertiary structures (Honda et al., 2004; Neidigh et al., 2002), yet have vital functions (e.g., subunits of F0- and F1-ATP synthases; Basrai et al., 1997; Kessler et al., 2003). Both extremes, in C. elegans , include highly conserved proteins.
For many years, working out protein homologies was done gene by individual gene (Swofford et al., 1996). While the techniques used were computational from the beginning, the data available for analysis were limited by the difficulty of manually isolating proteins and cloning genes. Wholesale genomic sequencing reversed this problem: there are now vast data available, but the expertise required to do manual phylogenetic analysis scales poorly to entire genomes, making methods for automatic protein phylogenetic analysis highly desirable.
One approach is to identify proteins as groups or clusters of homologs, leaving their orthology and paralogy undefined; this is how HMMs in PFAM and InterPro work. Homology groups can also be generated from BLAST searches of multiple genomes using Bayesian matrices (Enright et al., 2002). An advantage of homology-only searches is that they can dissect complex proteins into multiple domains easily; for instance, InterPro can mark each domain with an HMM corresponding to its pertinent family. A disadvantage is that such searches can lump proteins into large groups while ignoring their detailed evolutionary history. For instance, there is an InterPro family for protein kinases (IPR000719); however, this protein superfamily is multifarious, encompassing 134 orthologous families bound together by 8 ancient paralogies (Manning et al., 2002).
It would thus be highly desirable to have an agreed-upon way of constructing groups of orthologous and paralogous proteins, of the sort worked out for detecting homologous proteins by InterPro. Unfortunately, no such standard currently exists, though several strategies have been tried. Orthology groups were computed by Tatusov et al. (1997, 2003) who used triplets of mutual best BLAST hits to construct KOGs (euKaryotic Orthologous Groups), TWOGs (candidate TWo-species Orthologous Groups) and LSEs (Lineage-Specific Expansions peculiar to a single lineage) for two yeasts (Saccharomyces cerevisiae and Schizosaccharomyces pombe), one plant (Arabidopsis thaliana), and three metazoa (Homo sapiens, Drosophila melanogaster, and C. elegans). Remm et al. (2001) subsequently developed InParanoid, which generates pairwise orthologs and paralogs between pairs of species rather than several species at a time. Two methods of deriving orthology groups from PFAM have also been devised (HOPS and RIO: Storm and Sonnhammer, 2003; Zmasek and Eddy, 2002).
The classifications presented here, while useful, are necessarily imperfect. One challenge for an automatic classification is to correctly distinguish between protein motifs and protein families. Motifs are defined by InterPro as independent structural units that can be found either alone or with other domains or repeats, while families are defined as groups of proteins with shared domain or repeat architecture (Mulder et al., 2005). An InterPro motif can be present in a small set of C. elegans protein families, yet not exactly correspond with any one family. For example, the InterPro motif IPR007284 (DUF398/Ground-like domain) is found in both the groundhog (grd) and ground-like (grd) families, while the InterPro motifs IPR003586 and IPR003587 (Hint domains) are found in the groundhog and warthog (wrt) families, but none of these three motifs precisely identifies a gene family on its own (Aspöck et al., 1999). Another challenge is that C. elegans encodes gene families with remarkably high lineage-specific expansion and primary sequence divergence. Two well-studied instances of such families are seven-pass transmembrane receptors (Keating et al., 2003; Robertson, 1998, 2000, and 2001) and nuclear hormone receptors (Gissendanner et al. 2004; Maglich et al., 2001). In both cases, reliably sorting out the family members has absolutely required prolonged effort by experts; indeed, in the case of seven-pass receptors, classification is still going on (Chen et al., 2005; Thomas et al., 2005). Such protein families tend to be parceled out among InterPro and NCBI classes with limited accuracy.
By the WS130 archival release, WormBase had incorporated the PFAM/InterPro and NCBI KOG/TWOG/LSE families. In the near future, it is expected to also include InParanoid analyses. The rest of this chapter includes a summary of results from NCBI and PFAM/InterPro analyses.
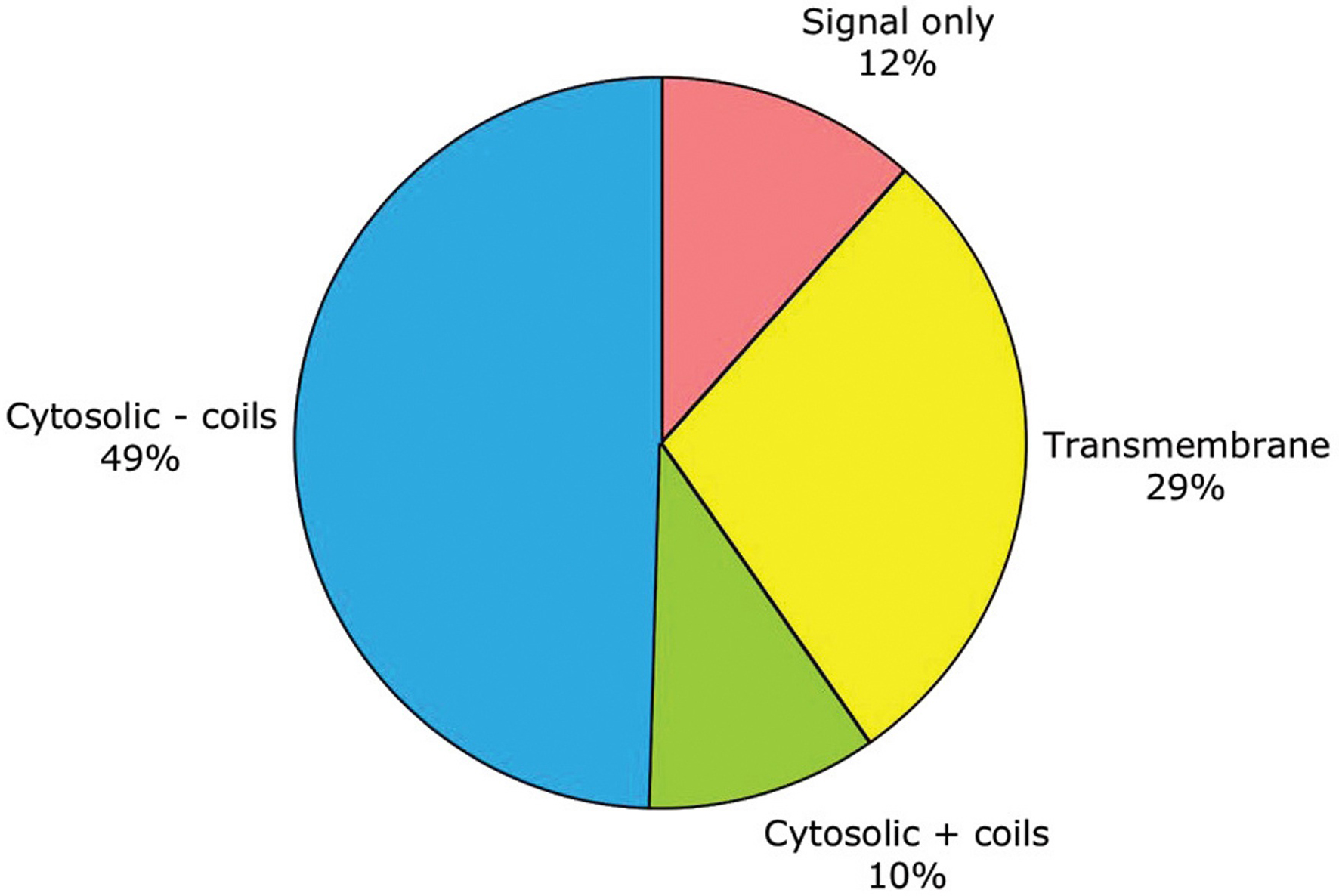
As noted above, subcellular localization can be roughly predicted from primary sequence. By this criterion, half of C. elegans genes encode purely cytosolic proteins, one-third encode membrane-embedded proteins, one-eighth encode secreted proteins, and one-tenth encode cytosolic proteins which aggregate through coiled-coils (see Figure 1; Table 1). No attempt has yet been made in WormBase to predict more fine-grained protein localization to the nucleus or other organelles.
Table 1. The numbers of C. elegans genes whose products are included in various groups. These are shown along with the number of protein families they encode
| Total genes | 19762 |
|---|---|
| Any family | 15544 |
| No family | 4218 |
| InterPro (IPR) | 5209 |
| KOG or TWOG | 10070 |
| KOG | 9515 |
| TWOG | 582 |
| LSE | 5321 |
| IPR and KOG/TWOG | 4208 |
| IPR, no KOG/TWOG | 1001 |
| KOG/TWOG, no IPR | 5862 |
| LSE only | 4473 |
| Signal only | 2303 |
| Transmembrane | 5683 |
| Cytosolic + coils | 1995 |
| Cytosolic - coils | 9781 |
| Total families | 6627 |
| IPR | 1337 |
| KOG | 4222 |
| TWOG | 225 |
| LSE | 843 |
|
Figure 1. General traits predicted for the products of 19,762 protein-coding genes. These traits include: having only a signal sequence predicted by SignalP (Nielsen and Krogh, 1998 ) but no transmembrane α-helices; having transmembrane α-helices predictedby TMHMM (Krogh et al., 2001 ); lacking either signal or transmembrane sequences (i.e., being putatively cytosolic); or being putatively cytosolic, but with one or more coiled-coil domains predicted by NCoils (Lupas, 1996).
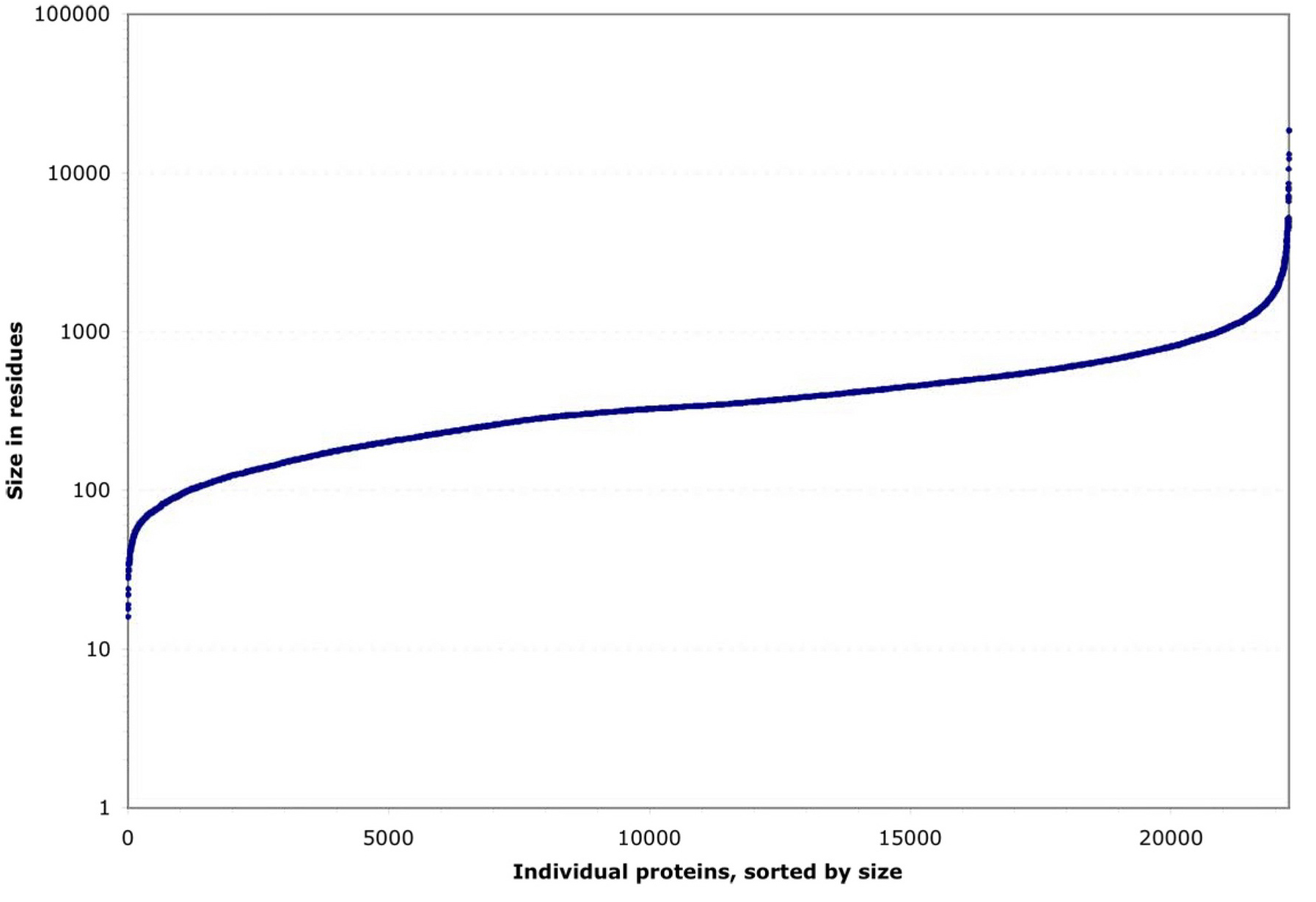
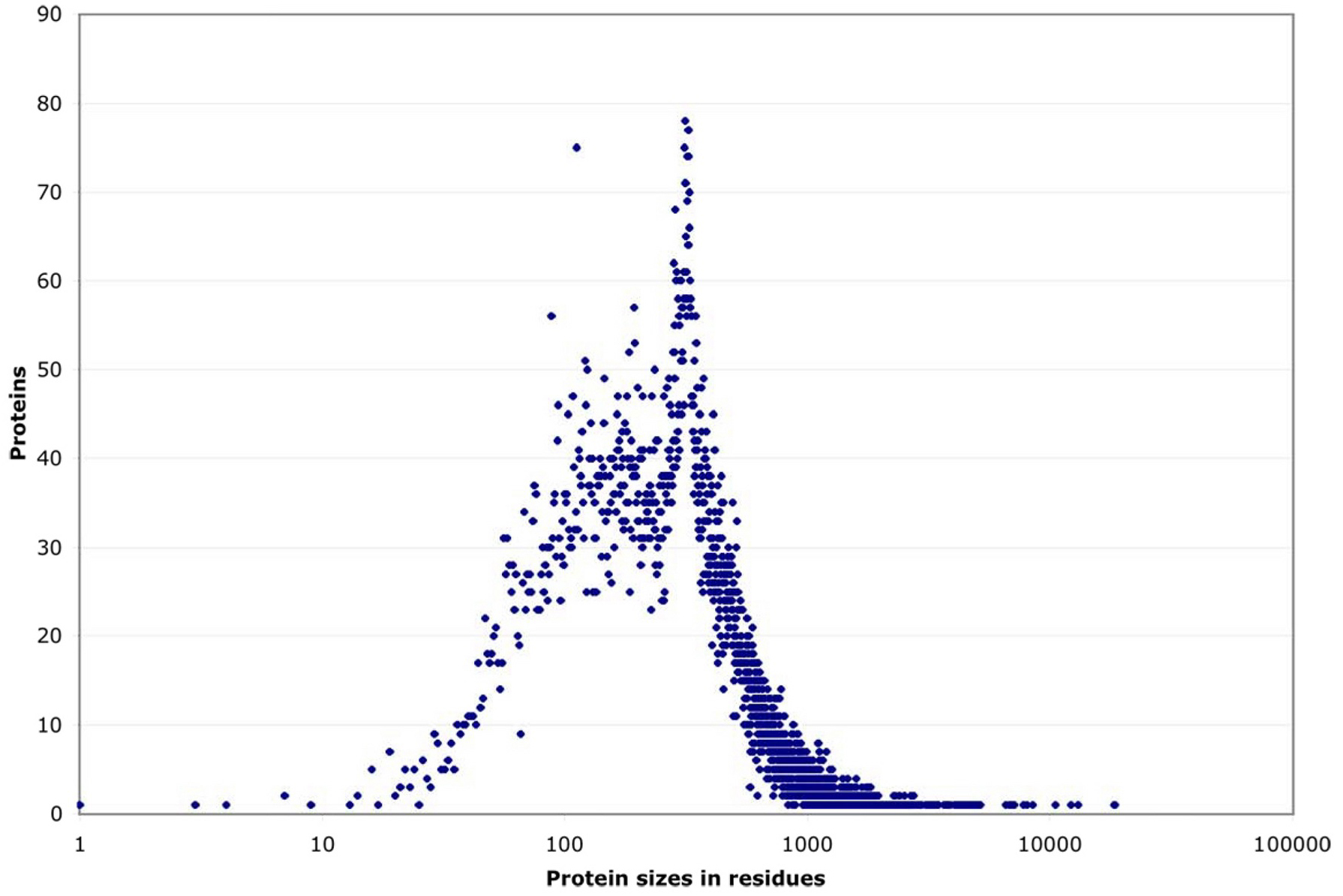
By their size, these proteins fall into three groups (see Figure 2). Roughly 90% have a strikingly regular logarithmic distribution of sizes from 100 to 1000 residues. This leaves two tails, each including ~5% of proteins, that deviate sharply downward or upward in size. Both the small and the large extremes include highly conserved proteins that probably cannot change size towards more normal levels without losing their function (e.g., small ribosomal proteins or large cytoskeletal ones). Within the central ~90% of proteins, most sizes are equally represented, except for a noticeable peak at ~340 residues caused by a nematode-specific expansion of chemosensory receptors (see Figure 3; Robertson, 1998; 2000 and 2001).
|
Figure 2. Residue length distribution for 22,246 individual C. elegans proteins. These are either known from cDNA sequences or predicted from genomic coding sequences.
|
Figure 3. Size distribution of proteins. Size distribution of proteins, listed by the number of proteins sharing each possible residue length. (In contrast, Figure 2 shows a series of individual proteins ascending in size.) Most proteins scatter broadly between ~80 and 1000 residues, but there is a noticeable peak at ~340 residues which corresponds to a large family of predicted 7-transmembrane receptors (Robertson, 2000).
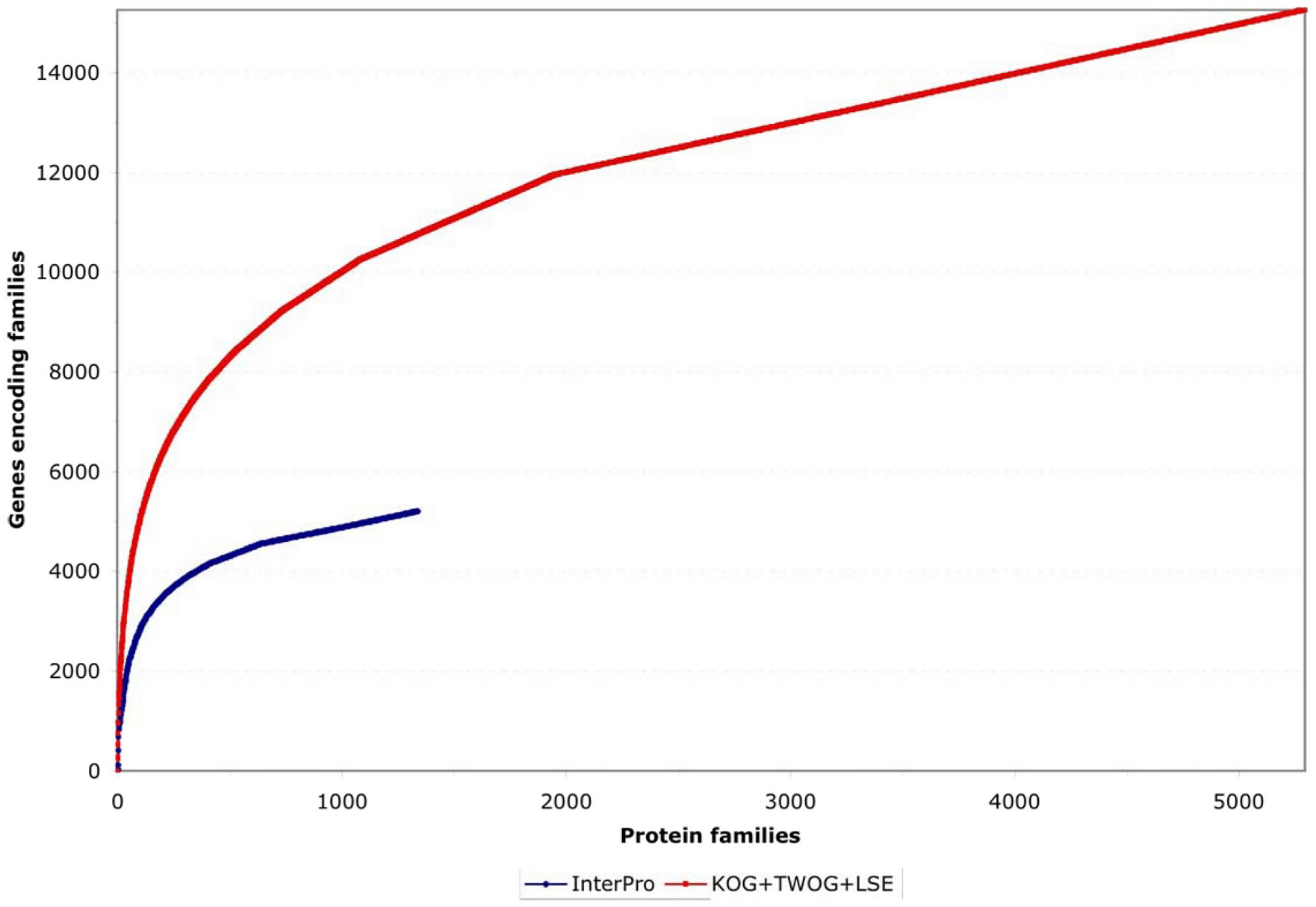
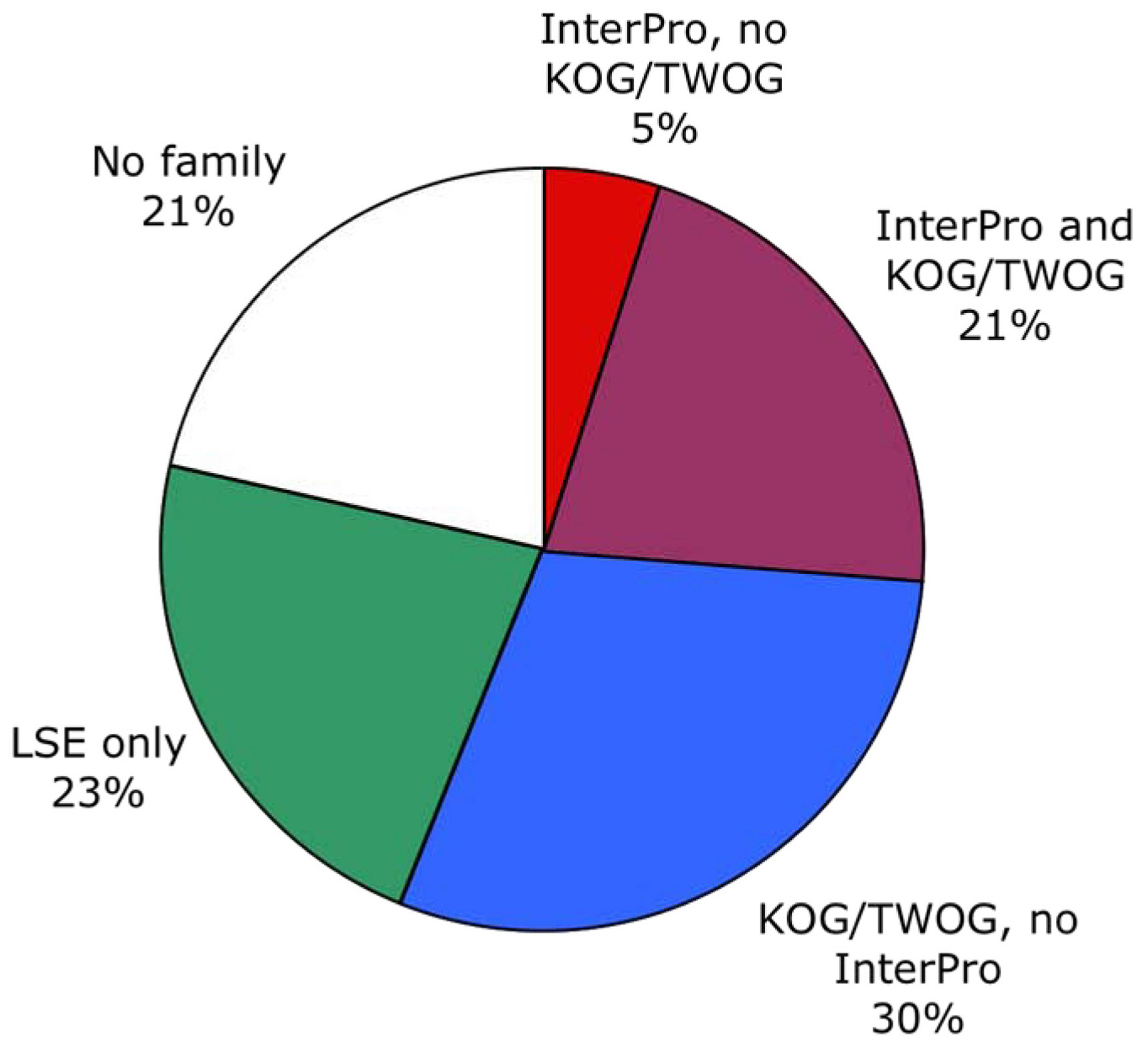
Two different systems of identifying protein families, from InterPro and NCBI, have been applied to C. elegans as of the WS130 release of WormBase. There are many different ways to examine these data, but one starting point is to look at how the protein families map to gene numbers (see Figure 4; Table 1). Both systems identify significant numbers of genes (~5000 and ~15,000), and both systems have some genes that they alone can identify (see Figure 5), but NCBI's families are considerably more extensive. Collectively, both methods provide some sort of identification for 79% of C. elegans protein-coding genes, going well beyond the functional classifications currently possible by mutant or RNAi phenotypes.
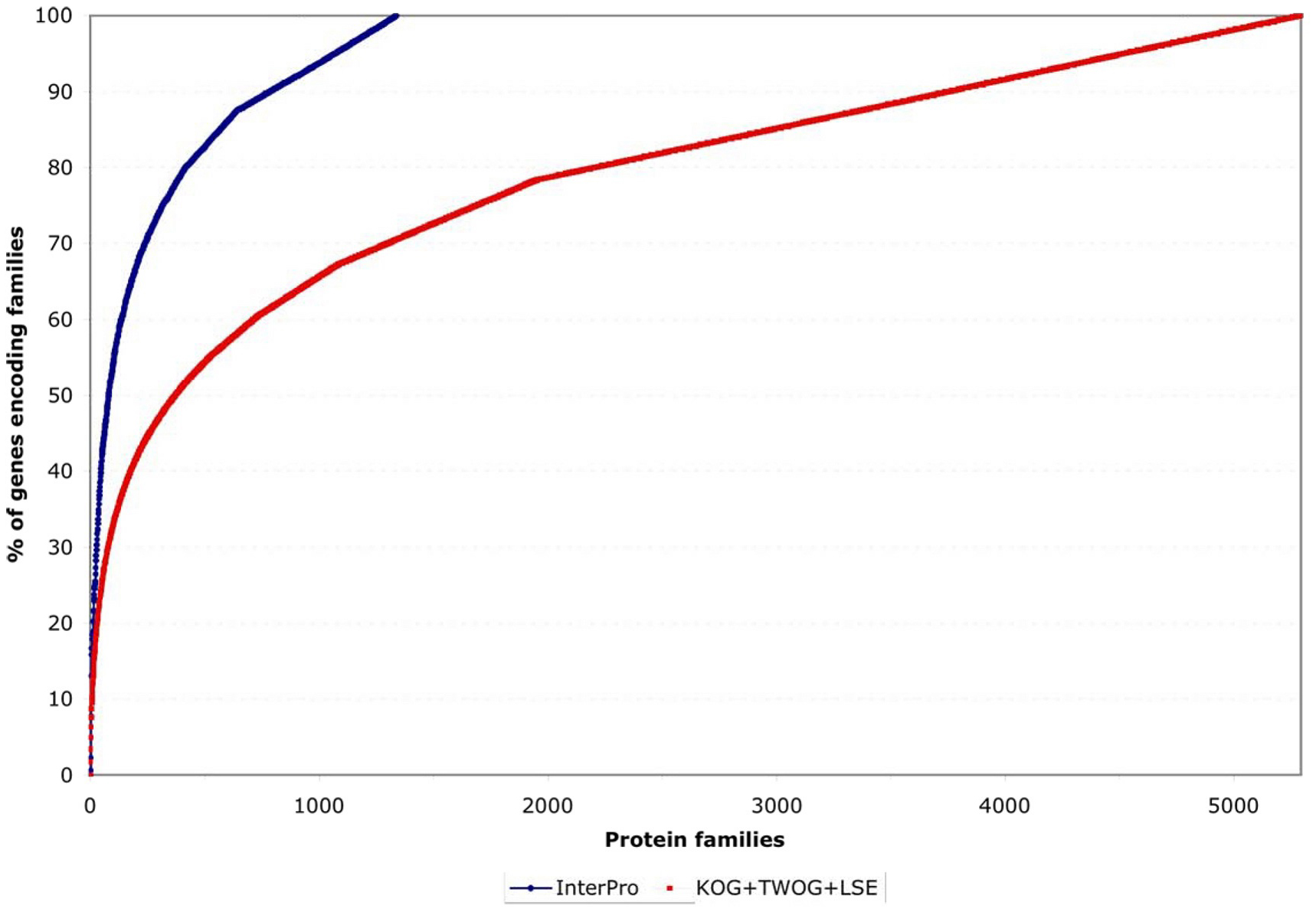
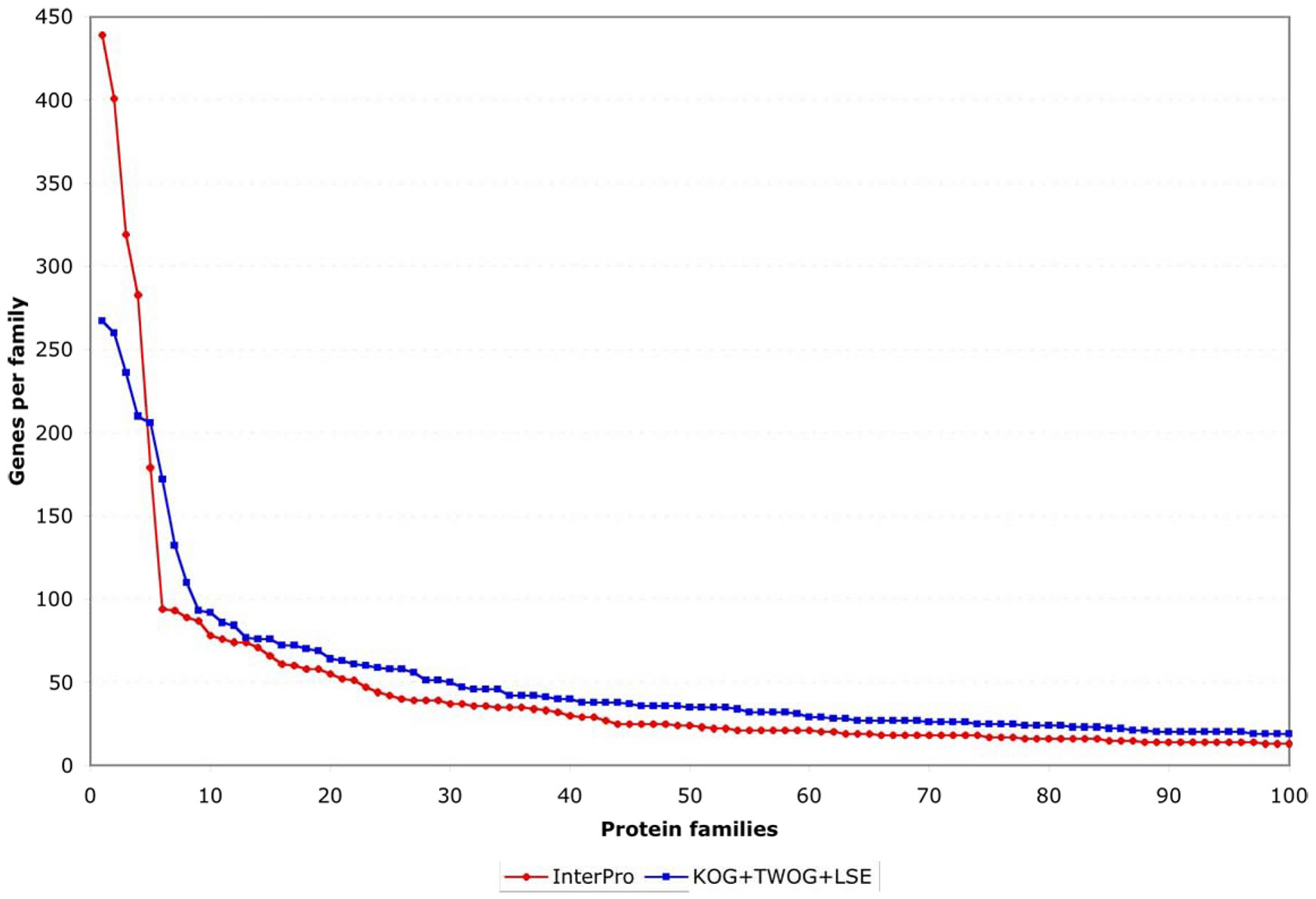
One striking feature of the protein family sets, whether from InterPro or from NCBI, is that they are very lopsided in how many genes individual families contain. This can be noticed by careful examination of Figure 4, but is easier to see if one plots the coverage of genes by protein families as a normalized curve (see Figure 6). Of all 5,209 genes with an InterPro family identification, 50% fall into only 80 families (out of 1,337 families) and 25% into only 21; of all 15,258 genes with an NCBI affiliation, 50% fall into only 372 NCBI families (out of 5,290 families) and 25% into only 51 (see Figure 6 and Figure 7; Tables 2 and 3). This is likely to reflect general trends of protein structural evolution, since 54 three-dimensional protein folds (6.6% of all folds) account for 76% of all known structures (Grant et al., 2004). In contrast, 699 InterPro and 3,341 NCBI families are encoded by only a single gene apiece in the C. elegans genome (see Figure 8).
|
Figure 4. Coverage of genes by protein families. The genes are sorted for maximum non-redundancy so that a gene falling into both a large and a small family is assigned to the small family; the results are then listed from the most common to the most uncommon families. Both InterPro and NCBI KOG/TWOG/LSE families cover significant numbers of genes, but the latter are more extensive.
|
Figure 5. Overall distribution of protein families among genes. Both InterPro and NCBI (KOG/TWOG/LSE) families are shown. There is significant overlap, but the NCBI families are more extensive. A fifth of C. elegans genes encode nematode-specific protein families (LSEs) of various types, while another fifth are not easily identifiable by homology.
|
Figure. 6. Normalized coverage of genes by families. This shows the same data as in Figure 4 but normalized to the total number of genes covered by the protein family set in question, making it clearer that a small number of protein families account for a disproportionate number of genes encompassed by a given family set (InterPro or NCBI).
|
Figure 7. Number of genes encoding members of the 100 most extensive InterPro or NCBI families. This shows that less than 10 families in either set are truly disproportionate in the number of genes encoding them (i.e., shoot far above a linear curve). However, the next 90 families, while also numerous, follow a more steadily declining distribution of sizes.
|
Figure 8. Frequency with which protein families are encoded by small or large numbers of genes. Both scales are logarithmic, to make the large variations easy to see. At one extreme, hundreds or thousands of families are encoded by only one gene apiece in the C. elegans genome. At the other extreme are instances of single protein families encoded by hundreds of genes.
Table 2. The most gene-populated InterPro protein families. These 80 families include 50% of all 5,209 genes encoding any InterPro family members. Full descriptions of these families, with references, are available at http://www.ebi.ac.uk/interpro (Mulder et al., 2005).
| Number of genes | InterPro Family |
|---|---|
| 439 | IPR000719 [Protein kinase] |
| 401 | IPR002290 [Serine/threonine protein kinase] |
| 319 | IPR001245 [Tyrosine protein kinase] |
| 283 | IPR003003 [7TM chemoreceptor, subfamily 2] |
| 179 | IPR000276 [Rhodopsin-like GPCR superfamily] |
| 94 | IPR000242 [Tyrosine specific protein phosphatase] |
| 93 | IPR000324 [Vitamin D receptor] |
| 89 | IPR006201 [Neurotransmitter-gated ion-channel] |
| 87 | IPR002198 [Short-chain dehydrogenase/reductase SDR] |
| 78 | IPR001128 [Cytochrome P450] |
| 76 | IPR001806 [Ras GTPase] |
| 74 | IPR002347 [Glucose/ribitol dehydrogenase] |
| 74 | IPR002401 [E-class P450, group I] |
| 71 | IPR003579 [Ras small GTPase, Rab type] |
| 66 | IPR002213 [UDP-glucuronosyl/UDP-glucosyltransferase] |
| 61 | IPR005821 [Ion transport protein] |
| 60 | IPR000169 [Peptidase, eukaryotic cysteine peptidase active site] |
| 58 | IPR003577 [Ras small GTPase, Ras type] |
| 58 | IPR000387 [Tyrosine specific protein phosphatase and dual specificity protein phosphatase] |
| 55 | IPR005828 [General substrate transporter] |
| 52 | IPR003578 [Ras small GTPase, Rho type] |
| 51 | IPR001534 [Transthyretin-like] |
| 47 | IPR001723 [Steroid hormone receptor] |
| 44 | IPR000609 [Integral membrane protein Srg, nematode type] |
| 42 | IPR003280 [K+ channel, two pore] |
| 40 | IPR003286 [Nematode reverse transcriptase-like] |
| 39 | IPR001506 [Peptidase M12A, astacin] |
| 39 | IPR006028 [Gamma-aminobutyric acid A receptor] |
| 39 | IPR004151 [C. elegans Sre G protein-coupled chemoreceptor] |
| 37 | IPR001993 [Mitochondrial substrate carrier] |
| 37 | IPR005829 [Sugar transporter superfamily] |
| 36 | IPR001283 [Allergen V5/Tpx-1 related] |
| 36 | IPR000344 [Nematode chemoreceptor, Sra] |
| 35 | IPR002041 [GTP-binding nuclear protein Ran] |
| 35 | IPR005834 [Haloacid dehalogenase-like hydrolase] |
| 35 | IPR001223 [Glycoside hydrolase, family 18] |
| 34 | IPR000718 [Peptidase M13, neprilysin] |
| 33 | IPR000215 [Proteinase inhibitor I4, serpin] |
| 32 | IPR000834 [Peptidase M14, carboxypeptidase A] |
| 30 | IPR008166 [Protein of unknown function DUF23] |
| 29 | IPR003392 [Patched] |
| 29 | IPR000668 [Peptidase C1A, papain] |
| 27 | IPR001873 [Na+ channel, amiloride-sensitive] |
| 25 | IPR001079 [Galectin, galactose-binding lectin] |
| 25 | IPR001394 [Peptidase C19, ubiquitin carboxyl-terminal hydrolase 2] |
| 25 | IPR002921 [Lipase, class 3] |
| 25 | IPR000615 [Bestrophin] |
| 25 | IPR000990 [Innexin] |
| 24 | IPR002492 [Transposase, Tc1/Tc3] |
| 24 | IPR001412 [Aminoacyl-tRNA synthetase, class I] |
| 23 | IPR000164 [Histone H3] |
| 22 | IPR001211 [Phospholipase A2] |
| 22 | IPR002659 [Glycosyl transferase, family 31] |
| 21 | IPR002394 [Nicotinic acetylcholine receptor] |
| 21 | IPR001019 [Guanine nucleotide binding protein [G-protein], alpha subunit] |
| 21 | IPR001757 [ATPase, E1-E2 type] |
| 21 | IPR006688 [ADP-ribosylation factor] |
| 21 | IPR001699 [Transcription factor, T-box] |
| 21 | IPR000566 [Lipocalin-related protein and Bos/Can/Equ allergen] |
| 20 | IPR003235 [Nematode insulin-related peptide, beta type] |
| 20 | IPR003406 [Glycosyl transferase, family 14] |
| 19 | IPR002119 [Histone H2A] |
| 19 | IPR002067 [Mitochondrial carrier protein] |
| 19 | IPR001461 [Peptidase A1, pepsin] |
| 18 | IPR002516 [Glycosyl transferase, family 11] |
| 18 | IPR000795 [Protein synthesis factor, GTP-binding] |
| 18 | IPR001163 [Small nuclear ribonucleoprotein [Sm protein]] |
| 18 | IPR002453 [Beta tubulin] |
| 18 | IPR002918 [Lipase, class 2] |
| 18 | IPR002113 [Adenine nucleotide translocator 1] |
| 18 | IPR001327 [FAD-dependent pyridine nucleotide-disulphide oxidoreductase] |
| 18 | IPR004825 [Insulin/IGF/relaxin] |
| 18 | IPR002184 [Integral membrane protein Srb, nematode type] |
| 17 | IPR000301 [CD9/CD37/CD63 antigen] |
| 17 | IPR000217 [Tubulin] |
| 17 | IPR000558 [Histone H2B] |
| 16 | IPR001951 [Histone H4] |
| 16 | IPR002123 [Phospholipid/glycerol acyltransferase] |
| 16 | IPR000910 [HMG1/2 [high mobility group] box] |
Table 3. The most gene-populated NCBI protein families. These 51 NCBI KOG/TWOG/LSE families include 25% of all 15,258 genes encoding any NCBI family members. NCBI's descriptions of these families are available at http://www.ncbi.nlm.nih.gov/COG/new/ (Tatusov et al., 1997 and 2003).
| Number of genes | NCBI KOG/TWOG/LSE family |
|---|---|
| 267 | LSE0262 [Predicted olfactory G-protein coupled receptor] |
| 260 | LSE0498 [7-transmembrane olfactory receptor] |
| 236 | KOG4297 [C-type lectin] |
| 210 | LSE0146 [Uncharacterized protein] |
| 206 | KOG3575 [Hormone receptors] |
| 172 | KOG3544 [Collagens (type IV and type XIII), and related proteins] |
| 132 | LSE0338 [Predicted transposase] |
| 110 | LSE0501 [7-transmembrane olfactory receptor] |
| 93 | KOG1164 [Casein kinase (serine/threonine/tyrosine protein kinase)] |
| 92 | LSE0150 [Uncharacterized protein, contains BTB/POZ domain] |
| 86 | KOG1721 [Zn-finger] |
| 84 | KOG3656 [7 transmembrane receptor] |
| 77 | LSE3897 [Unnamed protein] |
| 76 | LSE0021 [Sra family integral membrane protein] |
| 76 | KOG1192 [UDP-glucuronosyl and UDP-glucosyl transferase] |
| 72 | KOG1075 [Reverse transcriptase] |
| 72 | LSE0499 [Integral membrane O-acyltransferase] |
| 70 | LSE0023 [Predicted receptor] |
| 69 | LSE0147 [Chemoreceptor/7TM receptor] |
| 64 | LSE0503 [Secreted surface protein] |
| 63 | KOG0789 [Protein tyrosine phosphatase] |
| 61 | LSE0502 [7-transmembrane receptor] |
| 60 | LSE0562 [Nuclear hormone receptor] |
| 59 | KOG4735 [Extracellular protein with conserved cysteines] |
| 58 | LSE0504 [7-transmembrane receptor] |
| 58 | LSE0020 [Sre G protein-coupled chemoreceptor] |
| 56 | KOG2532 [Permease of the major facilitator superfamily] |
| 51 | LSE0018 [Uncharacterized protein] |
| 51 | LSE0505 [7-transmembrane receptor] |
| 50 | LSE0263 [F-box domain] |
| 47 | KOG3645 [Acetylcholine receptor] |
| 46 | KOG1418 [Tandem pore domain K+ channel] |
| 46 | KOG0156 [Cytochrome P450 CYP2 subfamily] |
| 46 | LSE0506 [Uncharacterized protein with conserved cysteine] |
| 42 | KOG1217 [Fibrillins and related proteins containing Ca2+-binding EGF-like domains] |
| 42 | LSE0508 [Uncharacterized protein] |
| 42 | KOG1695 [Glutathione S-transferase] |
| 41 | KOG1516 [Carboxylesterase and related proteins] |
| 40 | KOG0374 [Serine/threonine specific protein phosphatase PP1, catalytic subunit] |
| 40 | KOG3714 [Meprin A metalloprotease] |
| 38 | KOG0194 [Protein tyrosine kinase] |
| 38 | LSE0514 [Predicted secreted cysteine rich protein found only in C.elegans] |
| 38 | LSE0510 [Extracellular protein with cysteine rich structures] |
| 38 | LSE0509 [Uncharacterized protein] |
| 37 | LSE0511 [Uncharacterized protein] |
| 36 | KOG3017 [Defense-related protein containing SCP domain] |
| 36 | KOG0017 [Transposon-encoded proteins with TYA, reverse transcriptase, integrase domains in various combinations] |
| 36 | KOG2806 [Chitinase] |
| 36 | LSE0516 [Uncharacterized protein, contains major sperm protein (MSP) domain] |
| 35 | LSE0518 [7-transmembrane receptor] |
| 35 | KOG4185 [Predicted E3 ubiquitin ligase] |
By examining the membership of KOGs and TWOGs, it is possible to trace their origin to phylogenetic divisions between nematodes and other animal phyla, or between animals and other eukaryotes (Erwin and Davidson, 2002; King, 2004). There are 3951 KOGs shared by C. elegans, H. sapiens, and D. melanogaster; in contrast, the number of KOGs found in only two of these species is >10% of this number (331 KOGs in human and fly but not worm; 261 KOGs found in worm and human or fly). In some cases, a gene found in H. sapiens and D. melanogaster but not C. elegans may reflect simplication of the Caenorhabditis genome after the divergence of Caenorhabditis from other nematodes. For instance, orthologs of Hox3 and Antennapedia/Hox6 are missing from C. elegans but present in other nematodes (e.g., Brugia malayi; Aboobaker and Blaxter, 2003), as is the BRCA2-binding tumor suppressor EMSY (Hughes-Davies et al., 2003). Such genes may encode proteins that are needed for most metazoa but that have proven dispensable in the short-lived, anatomically minimal C. elegans and its close relatives. Evidence that loss of protein families may be a general trait of fast-breeding model organisms has recently come from EST sequencing of the staghorn coral Acropora millepora (Kortschak et al., 2003).
Some metazoan proteins which seem missing from C. elegans may actually be present, but be so divergent in their primary sequence that they are hard to recognize. Examples of such abnormally divergent proteins include the axin homolog PRY-1 (Korswagen et al., 2002), the BRCA1 ortholog BRC-1 (Boulton et al., 2004), the BRCA2 homolog BRC-2 (Bork et al., 1996), the opsin homolog SRO-1 (Troemel et al., 1995) the p53 ortholog CEP-1 (Derry et al., 2001; Schumacher et al., 2001), and the SKI/SNO homolog DAF-5 (da Graca et al. 2004) More generally, a higher divergence of many C. elegans proteins from those of H. sapiens versus D. melanogaster has been observed by Storm and Sonnhammer (2003), perhaps because nematodes are a deeply divergent phylum of the Coelomata (Wolf et al., 2004).
While gene loss and divergence tends to deplete C. elegans of recognizable protein families, other factors maintain or expand the population of C. elegans protein-coding genes. There is an overall tendency of complex eukaryotic genomes to have more paralogues than microbial genomes (Enright et al., 2003). One manifestation of this is for a gene family to differentially expand in a single metazoan phylum (e.g., nematodes). Such lineage-specific expansions were observed both by the KOG classification of NCBI and by Inparanoid; while a few of these expansions are shared by divergent phyla (such as arthropods and nematodes), they usually differ between phyla (Remm et al., 2001; Tatusov et al., 2003). Meanwhile, some gene families have been tenaciously retained from the origins of metazoa or eukaryotes until now: C. elegans shares 2518 KOGs with at least one species of unicellular eukaryotes, and shares 860 KOGs with six species of plants, animals, and unicellular eukaryotes (Soding and Lupas, 2003; Tatusov et al., 2003).
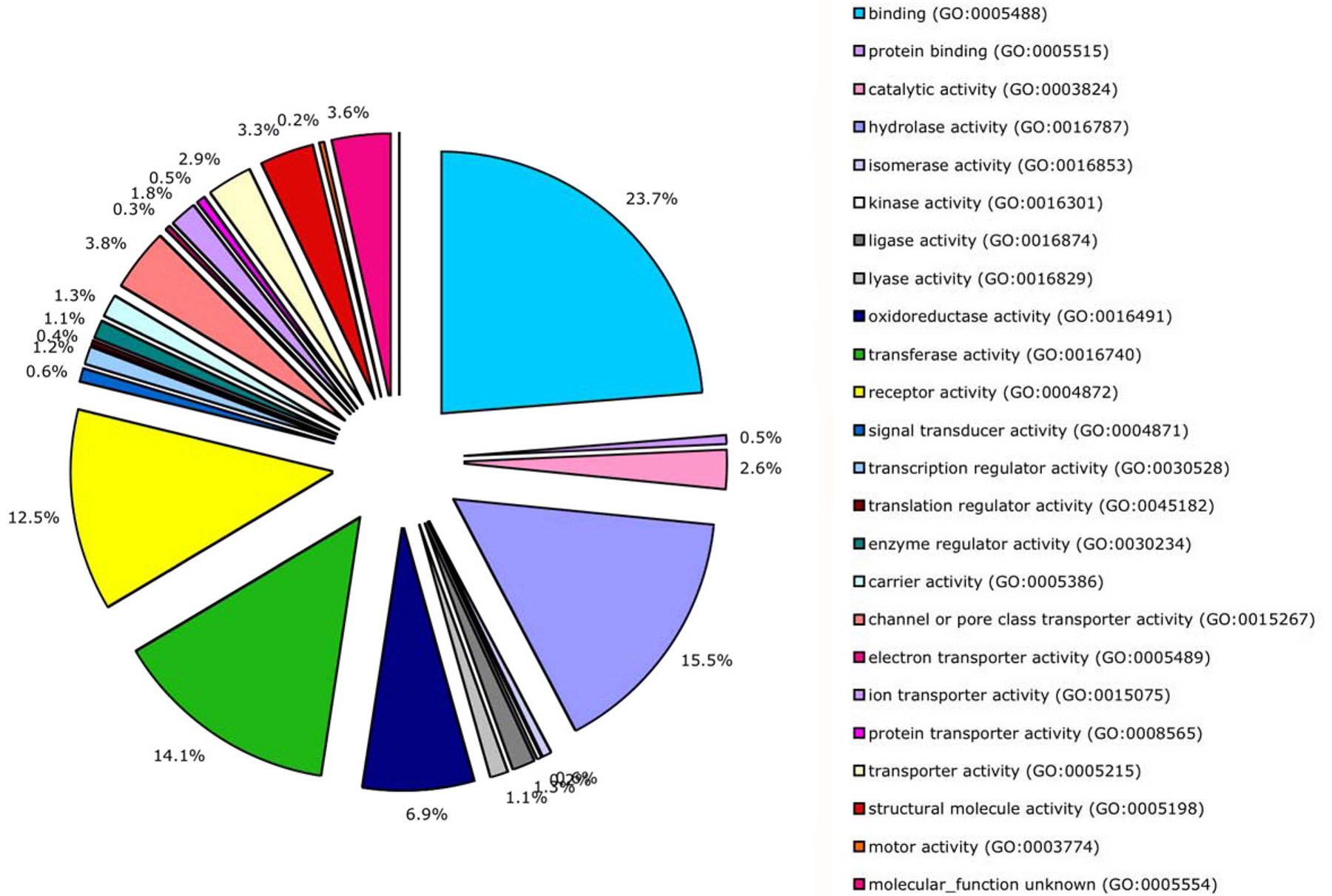
Both InterPro and NCBI protein families can be mapped to functional groups. For InterPro, the mapping involves correlating InterPro families with one or more terms in the Gene Ontology (GO) devised by Ashburner and coworkers (Gene Ontology Consortium, 2000; Camon et al., 2003). GO is a vocabulary for describing the functions of gene products, with three terminologies ("ontologies") specifying biochemical activity ("molecular function"), subcellular localization, and global biological purpose ("biological process"). A direct mapping of C. elegans genes to GO terms via InterPro families yields 333 different terms from the biological process ontology, and 556 from the molecular function one. These GO terms are lopsidedly distributed to C. elegans genes, mirroring the InterPro families from which they are derived (Tables 4 and 5). Because GO is extensive, abbreviated versions of GO ("GOslim") have been developed as aids to genome annotation (Camon et al., 2003; Gene Ontology Consortium, 2004). A summary of molecular function annotations with GOslim is shown in Figure 9; note that this only applies to the 26% of protein-coding genes that actually encode InterPro families. Over 50% of the inferred functions fall into three biochemical categories: binding of various ligands, hydrolysis, and molecular group transfers. Another ~20% fall into receptor activity or enzyme regulation. The remaining ~30% of GOslim annotations fall into 19 other categories.
Table 4. The most frequently predicted biological processes for InterPro families in C. elegans. These 33 biological process terms include 75% of all 4,753 gene annotations (for 4,062 genes), using GO terms derived from InterPro. Definitions of GO terms, with references, are available at http://geneontology.org (Gene Ontology Consortium, 2000).
| Number of genes | Biological process |
|---|---|
| 443 | protein amino acid phosphorylation (GO:0006468) |
| 339 | metabolism (GO:0008152) |
| 322 | proteolysis and peptidolysis (GO:0006508) |
| 213 | G-protein coupled receptor protein signaling pathway (GO:0007186) |
| 207 | transport (GO:0006810) |
| 205 | electron transport (GO:0006118) |
| 192 | regulation of transcription, DNA-dependent (GO:0006355) |
| 170 | ion transport (GO:0006811) |
| 163 | protein biosynthesis (GO:0006412) |
| 124 | protein amino acid dephosphorylation (GO:0006470) |
| 89 | intracellular protein transport (GO:0006886) |
| 86 | nucleosome assembly (GO:0006334) |
| 83 | protein transport (GO:0015031) |
| 79 | chromosome organization and biogenesis, sensu Eukaryota (GO:0007001) |
| 77 | small GTPase mediated signal transduction (GO:0007264) |
| 75 | sensory perception of chemical stimulus (GO:0007606) |
| 74 | carbohydrate metabolism (GO:0005975) |
| 69 | potassium ion transport (GO:0006813) |
| 66 | signal transduction (GO:0007165) |
| 45 | ubiquitin-dependent protein catabolism (GO:0006511) |
| 43 | sodium ion transport (GO:0006814) |
| 42 | lipid catabolism (GO:0016042) |
| 40 | RNA-dependent DNA replication (GO:0006278) |
| 39 | cation transport (GO:0006812) |
| 39 | tRNA aminoacylation for protein translation (GO:0006418) |
| 34 | lipid metabolism (GO:0006629) |
| 33 | amino acid metabolism (GO:0006520) |
| 32 | ATP synthesis coupled proton transport (GO:0015986) |
| 30 | DNA repair (GO:0006281) |
| 29 | physiological process (GO:0007582) |
| 29 | microtubule-based movement (GO:0007018) |
| 28 | protein amino acid glycosylation (GO:0006486) |
| 26 | response to oxidative stress (GO:0006979) |
Table 5. The most frequently predicted molecular functions for InterPro families in C. elegans. These 56 molecular function terms include 75% of all 7,343 gene annotations (for 4,846 genes), using GO terms derived from InterPro.
| Number of genes | Molecular function |
|---|---|
| 588 | ATP binding (GO:0005524) |
| 439 | protein kinase activity (GO:0004672) |
| 401 | protein serine/threonine kinase activity (GO:0004674) |
| 319 | protein-tyrosine kinase activity (GO:0004713) |
| 308 | DNA binding (GO:0003677) |
| 288 | G-protein coupled receptor activity (GO:0004930) |
| 218 | molecular function unknown (GO:0005554) |
| 185 | rhodopsin-like receptor activity (GO:0001584) |
| 172 | oxidoreductase activity (GO:0016491) |
| 155 | transporter activity (GO:0005215) |
| 141 | structural constituent of ribosome (GO:0003735) |
| 133 | transmembrane receptor activity (GO:0004888) |
| 132 | catalytic activity (GO:0003824) |
| 127 | GTP binding (GO:0005525) |
| 108 | ligand-dependent nuclear receptor activity (GO:0004879) |
| 101 | ion channel activity (GO:0005216) |
| 98 | protein tyrosine phosphatase activity (GO:0004725) |
| 92 | monooxygenase activity (GO:0004497) |
| 91 | extracellular ligand-gated ion channel activity (GO:0005230) |
| 87 | cysteine-type endopeptidase activity (GO:0004197) |
| 81 | hydrolase activity (GO:0016787) |
| 76 | RNA binding (GO:0003723) |
| 71 | transferase activity, transferring hexosyl groups (GO:0016758 |
| 58 | phosphoprotein phosphatase activity (GO:0004721) |
| 56 | binding (GO:0005488) |
| 53 | calcium ion binding (GO:0005509) |
| 47 | structural molecule activity (GO:0005198) |
| 46 | transcription factor activity (GO:0003700) |
| 42 | potassium channel activity (GO:0005267) |
| 40 | RNA-directed DNA polymerase activity (GO:0003964) |
| 39 | tRNA ligase activity (GO:0004812) |
| 39 | GABA-A receptor activity (GO:0004890) |
| 39 | astacin activity (GO:0008533) |
| 36 | serine-type endopeptidase inhibitor activity (GO:0004867) |
| 36 | signal transducer activity (GO:0004871) |
| 34 | cysteine-type peptidase activity (GO:0008234) |
| 34 | hormone activity (GO:0005179) |
| 34 | neprilysin activity (GO:0004245) |
| 32 | metalloendopeptidase activity (GO:0004222) |
| 32 | carboxypeptidase A activity (GO:0004182) |
| 30 | hydrogen-transporting ATP synthase activity, rotational mechanism (GO:0046933 |
| 29 | ubiquitin thiolesterase activity (GO:0004221) |
| 29 | hydrogen-transporting ATPase activity, rotational mechanism (GO:0046961) |
| 29 | hedgehog receptor activity (GO:0008158) |
| 28 | lipid binding (GO:0008289) |
| 27 | sodium channel activity (GO:0005272) |
| 27 | peroxidase activity (GO:0004601) |
| 27 | acyltransferase activity (GO:0008415) |
| 25 | sugar binding (GO:0005529) |
| 25 | triacylglycerol lipase activity (GO:0004806) |
| 24 | chaperone activity (GO:0003754) |
| 24 | transposase activity (GO:0004803) |
| 23 | zinc ion binding (GO:0008270) |
| 22 | galactosyltransferase activity (GO:0008378) |
| 22 | phospholipase A2 activity (GO:0004623) |
| 21 | nicotinic acetylcholine-activated cation-selective channel activity (GO:0004889) |
|
Figure 9. Overview of protein-encoding gene functions. Overview of protein-encoding gene functions, as summarized by InterPro families and their mapping to molecular function terms. This mapping uses a GOslim developed by the EBI for annotating entire proteomes in SwissProt (Camon et al., 2003).
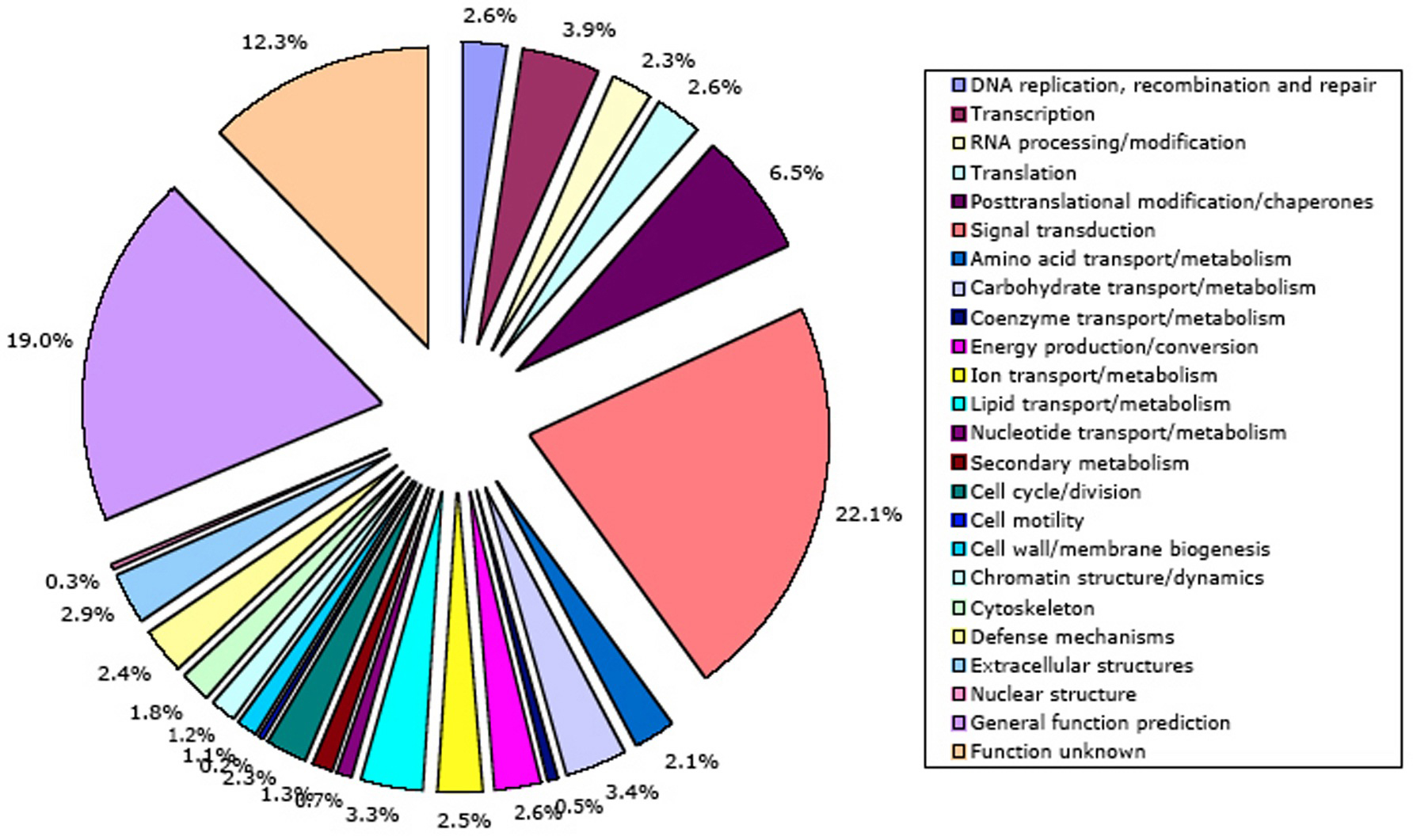
Independently, NCBI KOG/TWOG/LSE families have been placed in 24 functional categories by Koonin and coworkers (Tatusov et al., 2003). A mapping of C. elegans genes to these categories is shown in Figure 10. The NCBI classification has not yet been mapped onto GO, a much more structured and widely used system. However, NCBI functional annotations currently cover more of the C. elegans genome (66% of protein-coding genes) than InterPro annotations. The single well-defined function that summarizes a truly disproportionate fraction of NCBI gene annotations (22.1%) is signal transduction; 21 other functional annotations all get much smaller sets of genes (0.2-6.5% apiece). 19% of genes have only a broad guess at their function, and 12% of genes are functionally unknown.
This work was supported by a grant (# P41 HG02223) from theNational Human Genome Research Institute at the U.S. National Institutes of Health. I thank Darin Blasiar and Kimberly Van Auken for computing the InterPro search results againstWormBase release WS130, and my colleagues in WormBase for providing the scientific environment that allowed this chapter to be written. I thank Titus Brown, Karin Kiontke, Paul Sternberg, Weiwei Zhong, and two anonymous reviewers for helpful comments on the manuscript.
Aboobaker, A.A., and Blaxter, M.L. (2003). Hox gene loss during dynamic evolution of the nematode cluster. Curr. Biol. 13, 37–40. Abstract Article
Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. Abstract Article
Aspöck, G., Kagoshima, H., Niklaus, G., and Bürglin, T.R. (1999). Caenorhabditis elegans has scores of hedgehog-related genes: sequence and expression analysis. Genome Res. 9, 909–923. Abstract Article
Aspöck, G., Ruvkun, G., and Bürglin, T.R. (2003). The Caenorhabditis elegans ems class homeobox gene ceh-2 is required for M3 pharynx motoneuron function. Development 130, 3369–3378. Abstract Article
Basrai, M.A., Hieter, P., and Boeke, J.D. (1997). Small open reading frames: beautiful needles in the haystack. Genome Res. 7, 768–771. Abstract
Bateman, A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L., et al. (2004). The Pfam protein families database. Nucleic Acids Res. 32, D138–D141. Abstract Article
Bork, P., Blomberg, N., and Nilges, M. (1996). Internal repeats in the BRCA2 protein sequence. Nat. Genet. 13, 22–23. Abstract Article
Boulton, S.J., Martin, J.S., Polanowska, J., Hill, D.E., Gartner, A., and Vidal, M. (2004). BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr. Biol. 14, 33–39. Abstract Article
Camon, E., Magrane, M., Barrell, D., Binns, D., Fleischmann, W., Kersey, P., Mulder, N., Oinn, T., Maslen, J., Cox, A., et al. (2003). The Gene Ontology Annotation (GOA) project: implementation of GO in SWISS-PROT, TrEMBL, and InterPro. Genome Res. 13, 662–672. Abstract Article
Chance, M.R., Fiser, A., Sali, A., Pieper, U., Eswar, N., Xu, G., Fajardo, J.E., Radhakannan, T., and Marinkovic, N. (2004). High-throughput computational and experimental techniques in structural genomics. Genome Res. 14, 2145–2154. Abstract Article
Chen, N., Pai, S., Zhao, Z., Mah, A., Newbury, R., Johnsen, R.C., Altun, Z., Moerman, D.G., Baillie, D.L., and Stein, L.D. (2005). Identification of a nematode chemosensory gene family. Proc. Natl. Acad. Sci. U.S.A. 102, 146–151. Abstract Article
Colaiacovo, M.P., Stanfield, G.M., Reddy, K.C., Reinke, V., Kim, S.K., and Villeneuve, A.M. (2002). A targeted RNAi screen for genes involved in chromosome morphogenesis and nuclear organization in the Caenorhabditis elegans germline. Genetics 162, 113–128. Abstract
da Graca, L.S., Zimmerman, K.K., Mitchell, M.C., Kozhan-Gorodetska, M., Sekiewicz, K., Morales, Y., and Patterson, G.I. (2004). DAF-5 is a Ski oncoprotein homolog that functions in a neuronal TGF-β pathway to regulate C. elegans dauer development. Development 131, 435–446. Abstract Article
De Beer, G. (1997). Homology: an unsolved problem. In: Evolution, M. Ridley, ed. (New York: Oxford University Press), pp. 213–221.
Derry, W.B., Putzke, A.P., and Rothman, J.H. (2001). Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294, 591–595. Abstract Article
Duerr, J.S., Frisby, D.L., Gaskin, J., Duke, A., Asermely, K., Huddleston, D., Eiden, L.E., and Rand, J.B. (1999). The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J. Neurosci. 19, 72–84. Abstract
Durbin, R., Eddy, S.R., Krogh, A., and Mitchison, G. (1999). Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids (Cambridge, UK: Cambridge University Press).
Dyson, H.J., and Wright, P.E. (2005). Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208. Abstract Article
Eddy, S.E. (2005). http://hmmer.wustl.edu.
Enright, A.J., Kunin, V., and Ouzounis, C.A. (2003). Protein families and TRIBES in genome sequence space. Nucleic Acids Res. 31, 4632–4638. Abstract Article
Enright, A.J., Van Dongen, S., and Ouzounis, C.A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. Abstract Article
Erwin, D.H., and Davidson, E.H. (2002). The last common bilaterian ancestor. Development 129, 3021–3032. Abstract
Fay, J.C., and Wu, C.I. (2003). Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genomics Hum. Genet. 4, 213–235. Abstract Article
Fitch, W.M. (1970). Distinguishing homologous from analogous proteins. Syst. Zool. 19, 99–113. Abstract
Fitch, W.M. (2000). Homology a personal view on some of the problems. Trends Genet. 16, 227–231. Abstract Article
Galperin, M.Y., Walker, D.R., and Koonin, E.V. (1998). Analogous enzymes: independent inventions in enzyme evolution. Genome Res. 8, 779–790. Abstract
Gene Ontology Consortium (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. Abstract Article
Gene Ontology Consortium (2004). The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261. Abstract Article
Gerlt, J.A., and Babbitt, P.C. (2001). Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu. Rev. Biochem. 70, 209–246. Abstract Article
Gissendanner, C.R., Crossgrove, K., Kraus, K.A., Maina, C.V., and Sluder, A.E. (2004). Expression and function of conserved nuclear receptor genes in Caenorhabditis elegans. Dev. Biol. 266, 399–416. Abstract Article
Grant, A., Lee, D., and Orengo, C. (2004). Progress towards mapping the universe of protein folds. Genome Biol. 5, 107. Abstract Article
Gray, G.S., and Fitch, W.M. (1983). Evolution of antibiotic resistance genes: the DNA sequence of a kanamycin resistance gene from Staphylococcus aureus. Mol. Biol. Evol. 1, 57–66. Abstract
Gribskov, M., Luthy, R., and Eisenberg, D. (1990). Profile analysis. Methods Enzymol. 183, 146–159. Abstract
Haun, C., Alexander, J., Stainier, D.Y., and Okkema, P.G. (1998). Rescue of Caenorhabditis elegans pharyngeal development by a vertebrate heart specification gene. Proc. Natl. Acad. Sci. U.S.A. 95, 5072–5075. Abstract Article
Honda, S., Yamasaki, K., Sawada, Y., and Morii, H. (2004). 10 residue folded peptide designed by segment statistics. Structure 12, 1507–1518. Abstract Article
Hughes-Davies, L., Huntsman, D., Ruas, M., Fuks, F., Bye, J., Chin, S.F., Milner, J., Brown, L.A., Hsu, F., Gilks, B., et al. (2003). EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 115, 523–535. Abstract Article
Hutter, H., Vogel, B.E., Plenefisch J.D., Norris C.R., Proenca R.B., Spieth J., Guo C., Mastwal S., Zhu X., Scheel J., et al. (2000). Conservation and novelty in the evolution of cell adhesion and extracellular matrix genes. Science 287, 989–994. Abstract Article
Huyen, Y., Jeffrey, P.D., Derry, W.B., Rothman, J.H., Pavletich, N.P., Stavridi, E.S., and Halazonetis, T.D. (2004). Structural differences in the DNA binding domains of human p53 and its C. elegans ortholog Cep-1. Structure 12, 1237–1243. Abstract Article
Käll, L., Krogh, A., and Sonnhammer, E.L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036. Abstract Article
Karlin, S., Brocchieri, L., Bergman, A., Mrazek, J., and Gentles, A.J. (2002). Amino acid runs in eukaryotic proteomes and disease associations. Proc. Natl. Acad. Sci. U.S.A. 99, 333–338. Abstract Article
Keating, C.D., Kriek, N., Daniels, M., Ashcroft, N.R., Hopper, N.A., Siney, E.J., Holden-Dye, L., and Burke, J.F. (2003). Whole-genome analysis of 60 G protein-coupled receptors in Caenorhabditis elegans by gene knockout with RNAi. Curr. Biol. 13, 1715–1720. Abstract Article
Kessler, M.M., Zeng, Q., Hogan, S., Cook, R., Morales, A.J., and Cottarel, G. (2003). Systematic discovery of new genes in the Saccharomyces cerevisiae genome. Genome Res. 13, 264–271. Abstract Article
King, N. (2004). The unicellular ancestry of animal development. Dev. Cell 7, 313–325. Abstract Article
Korswagen, H.C., Coudreuse, D.Y., Betist, M.C., van de Water, S., Zivkovic, D., and Clevers, H.C. (2002). The Axin-like protein PRY-1 is a negative regulator of a canonical Wnt pathway in C. elegans. Genes Dev. 16, 1291–1302. Abstract Article
Kortschak, R.D., Samuel, G., Saint, R., and Miller, D.J. (2003). EST analysis of the cnidarian Acropora millepora reveals extensive gene loss and rapid sequence divergence in the model invertebrates. Curr. Biol. 13, 2190–2195. Abstract Article
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E.L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. Abstract Article
Kurland, C.G., Canback, B., and Berg, O.G. (2003). Horizontal gene transfer: a critical view. Proc. Natl. Acad. Sci. U.S.A. 100, 9658–9662. Abstract Article
Lee, R.Y., Hench, J., and Ruvkun, G. (2001). Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr. Biol. 11, 1950–1957. Abstract Article
Lee, J., Jongeward, G.D., and Sternberg, P.W. (1994). unc-101, a gene required for many aspects of Caenorhabditis elegans development and behavior, encodes a clathrin-associated protein. Genes Dev. 8, 60–73. Abstract
Levitan, D., Doyle, T.G., Brousseau, D., Lee, M.K., Thinakaran, G., Slunt, H.H., Sisodia, S.S., and Greenwald, I. (1996). Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 93, 14940–14944. Abstract Article
Liu, J., Tan, H., and Rost, B. (2002). Loopy proteins appear conserved in evolution. J. Mol. Biol. 322, 53–64. Abstract Article
Luan, C.H., Qiu, S., Finley, J.B., Carson, M., Gray, R.J., Huang, W., Johnson, D., Tsao, J., Reboul, J., Vaglio, P., et al. (2004). High-throughput expression of C. elegans proteins. Genome Res. 14, 2102–2110. Abstract Article
Lupas, A. (1996). Prediction and analysis of coiled-coil structures. Methods Enzymol. 266, 513–525. Abstract
Maglich, J.M., Sluder, A., Guan, X., Shi, Y., McKee, D.D., Carrick, K., Kamdar, K., Willson, T.M., and Moore, J.T. (2001). Comparison of complete nuclear receptor sets from the human, Caenorhabditis elegans and Drosophila genomes. Genome Biol. 2, RESEARCH0029. Abstract
Manning, G., Whyte, D.B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. Abstract Article
Marchler-Bauer, A., Anderson, J.B., Cherukuri, P.F., DeWeese-Scott, C., Geer, L.Y., Gwadz, M., He, S., Hurwitz, D.I., Jackson, J.D., Ke, Z., et al. (2005). CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 33, D192–D196. Abstract Article
Meng, E.C., Polacco, B.J., and Babbitt, P.C. (2004). Superfamily active site templates. Proteins 55, 962–976. Abstract Article
Michelitsch, M.D., and Weissman, J.S. (2000). A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. U.S.A. 97, 11910–11915. Abstract Article
Morett, E., Korbel, J.O., Rajan, E., Saab-Rincon, G., Olvera, L., Olvera, M., Schmidt, S., Snel, B., and Bork, P. (2003). Systematic discovery of analogous enzymes in thiamin biosynthesis. Nat. Biotechnol. 21, 790–795. Abstract Article
Moult, J., Fidelis, K., Zemla, A., and Hubbard, T. (2003). Critical assessment of methods of protein structure prediction (CASP)-round V. Proteins 53, 334–339. Abstract Article
Mulder, N.J., Apweiler, R., Attwood, T.K., Bairoch, A., Bateman, A., Binns, D., Bradley, P., Bork, P., Bucher, P., Cerutti, L., et al. (2005). InterPro, progress and status in 2005. Nucleic Acids Res. 33, D201–D205. Abstract Article
Nair, R., and Rost, B. (2004). LOCnet and LOCtarget: sub-cellular localization for structural genomics targets. Nucleic Acids Res. 32, W517–W521. Abstract
Neidigh, J.W., Fesinmeyer, R.M., and Andersen, N.H. (2002). Designing a 20-residue protein. Nat. Struct. Biol. 9, 425–430. Abstract Article
Nielsen, H., and Krogh, A. (1998). Prediction of signal peptides and signal anchors by a hidden Markov model. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 122–130. Abstract
Ouzounis, C.A., Coulson, R.M., Enright, A.J., Kunin, V., and Pereira-Leal, J.B. (2003). Classification schemes for protein structure and function. Nat. Rev. Genet. 4, 508–519. Abstract Article
Park, K.J., and Kanehisa, M. (2003). Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics 19, 1656–1663. Abstract Article
Pierce, K.L., Premont, R.T., and Lefkowitz, R.J. (2002). Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650. Abstract Article
Promponas, V.J., Enright, A.J., Tsoka, S., Kreil, D.P., Leroy, C., Hamodrakas, S., Sander, C., and Ouzounis, C.A. (2000). CAST: an iterative algorithm for the complexity analysis of sequence tracts. Bioinformatics 16, 915–922. Abstract Article
Remm, M., Storm, C.E., and Sonnhammer, E.L. (2001). Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 314, 1041–1052. Abstract Article
Robertson, H.M. (1998). Two large families of chemoreceptor genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae reveal extensive gene duplication, diversification, movement, and intron loss. Genome Res. 8, 449–463. Abstract
Robertson, H.M. (2000). The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 10, 192–203. Abstract Article
Robertson, H.M. (2001). Updating the str and srj(stl) families of chemoreceptors in Caenorhabditis nematodes reveals frequent gene movement within and between chromosomes. Chem. Senses 26, 151–159. Abstract Article
Scholl, E.H., Thorne, J.L., McCarter, J.P., and Bird, D.M. (2003). Horizontally transferred genes in plant-parasitic nematodes: a high-throughput genomic approach. Genome Biol. 4, R39. Abstract Article
Schumacher, B., Hofmann, K., Boulton, S., and Gartner, A. (2001). The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 11, 1722–1727. Abstract Article
Scott, M.S., Thomas, D.Y., and Hallett, M.T. (2004). Predicting subcellular localization via protein motif co-occurrence. Genome Res. 14, 1957–1966. Abstract Article
Si, K., Lindquist, S., and Kandel, E.R. (2003). A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 115, 879–891. Abstract Article
Siew, N., and Fischer, D. (2004). Structural biology sheds light on the puzzle of genomic ORFans. J. Mol. Biol. 342, 369–373. Abstract Article
Soding, J., and Lupas, A.N. (2003). More than the sum of their parts: on the evolution of proteins from peptides. Bioessays 25, 837–346. Abstract Article
Solari, F., Bourbon-Piffaut, A., Masse, I., Payrastre, B., Chan, A.M., and Billaud, M. (2005). The human tumour suppressor PTEN regulates longevity and dauer formation in Caenorhabditis elegans. Oncogene 24, 20–27. Abstract Article
Sonnhammer, E.L., and Koonin, E.V. (2002). Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 18, 619–620. Abstract Article
Stein, L.D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M.R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al. (2003). The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1, E45. Abstract Article
Storm, C.E., and Sonnhammer, E.L. (2003). Comprehensive analysis of orthologous protein domains using the HOPS database. Genome Res. 13, 2353–2362. Abstract Article
Swofford, D.L., Olsen, G.J., Waddell, P.J., and Hillis, D.M. (1996). Phylogenetic inference. In: Molecular Systematics, 2nd edn, D.M. Hillis, C. Moritz, and B.K. Mable, eds. (Sunderland, MA: Sinauer Associates, Inc.), Pp. 407–514.
Tatusov, R.L., Fedorova, N.D., Jackson, J.D., Jacobs, A.R., Kiryutin, B., Koonin, E.V., Krylov, D.M., Mazumder, R., Mekhedov, S.L., Nikolskaya, A.N., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41. Abstract Article
Tatusov, R.L., Koonin, E.V., and Lipman, D.J. (1997). A genomic perspective on protein families. Science 278, 631–637. Abstract Article
Taylor, J.S., and Raes, J. (2004). Duplication and divergence: the evolution of new genes and old ideas. Annu. Rev. Genet. 38, 615–643. Abstract Article
Thomas, J.H., Kelley, J.L., Robertson, H.M., Ly, K., and Swanson, W.J. (2005). Adaptive evolution in the SRZ chemoreceptor families of Caenorhabditis elegans and Caenorhabditis briggsae. Proc. Natl. Acad. Sci. U.S.A. 102, 4476–4481. Abstract Article
Troemel, E.R., Chou, J.H., Dwyer, N.D., Colbert, H.A., and Bargmann, C.I. (1995). Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell 83, 207–218. Abstract Article
Wan, H., Li, L., Federhen, S., and Wootton, J.C. (2003). Discovering simple regions in biological sequences associated with scoring schemes. J. Comput. Biol. 10, 171–185. Abstract Article
Westmoreland, J.J., McEwen, J., Moore, B.A., Jin, Y., and Condie, B.G. (2001). Conserved function of Caenorhabditis elegans UNC-30 and mouse Pitx2 in controlling GABAergic neuron differentiation. J. Neurosci. 21, 6810–6819. Abstract
Wolf, Y.I., Rogozin, I.B., and Koonin E.V. (2004). Coelomata and not Ecdysozoa: evidence from genome-wide phylogenetic analysis. Genome Res. 14, 29–36. Abstract Article
*Edited by Jonathan Hodgkin and Philip Anderson. Last revised April 02, 2005. Published September 23, 2005. This chapter should be cited as: Schwarz, E.M. Genomic classification of protein-coding gene families (September 23, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.29.1, http://www.wormbook.org.
Copyright: © 2005 Erich M. Schwarz. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: emsch@its.caltech.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.