Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergAbstract
A genetic enhancer is a mutation in one gene that intensifies the phenotype caused by a mutation in another gene. The phenotype of the double mutant is much stronger than the summation of the single mutant phenotypes. The isolation of enhancers can lead to the identification of interacting genes, including genes that act redundantly with respect to each other. Examples in Caenorhabditis elegans of dominant enhancers are presented first, followed by a review of recessive enhancers of null mutations. In some of these cases, the interacting genes are related in structure and function, but in other cases, the interacting genes are nonhomologous. Recessive enhancers of non-null mutations can also be useful. A powerful advance for the identification of recessive enhancers is genome-wide screening based on RNA interference.
A mutation in one gene that enhances or exacerbates the phenotype caused by a mutation in another gene is called an enhancer (not be confused with an enhancer DNA element, which is a cis-acting regulator of transcription). The enhancement must exceed what one would expect from a simple summation of the phenotypic effects of the two mutations on their own, and thus each mutation can be considered to be an enhancer of the other. When the double mutant exhibits a phenotype not apparent in either single mutant, the novel phenotype is referred to as synthetic. Although one should consider the possibility that mutations in two genes that have completely unrelated functions might overburden an organism and be unexpectedly deleterious even though the single mutants are relatively healthy, the identification of enhancers in C. elegans has in practice often led to the discovery of genes that interact in informative ways. Enhancement may arise from different types of genetic interaction. For example, two genes may provide a specific function redundantly, in which case the homozygous double mutant may exhibit a much stronger phenotype than either single mutant. Alternatively, mutations in two genes that are both required for the same function, say by encoding either components of a protein complex or different steps in a signal transduction pathway, may synergize and lead to an enhanced double mutant phenotype.
For two genes that exhibit enhancement, insight into the possible type of genetic interaction can sometimes be gleaned from the nature of the mutations affecting the two genes. For example, null mutations that enhance each other identify overlapping or partially redundant functions. Each mutation on its own may cause a phenotype, but the double mutant exhibits either a novel or a much enhanced phenotype relative to either single mutant (Figure 1). The overlapping functions may be homologous, or they may be independent functions that affect a specific biological process redundantly. Many important functions appear to be provided redundantly or at least partially redundantly, and screens for enhancement of null mutations are likely to become increasingly important for the elucidation of worm development and behavior.
|
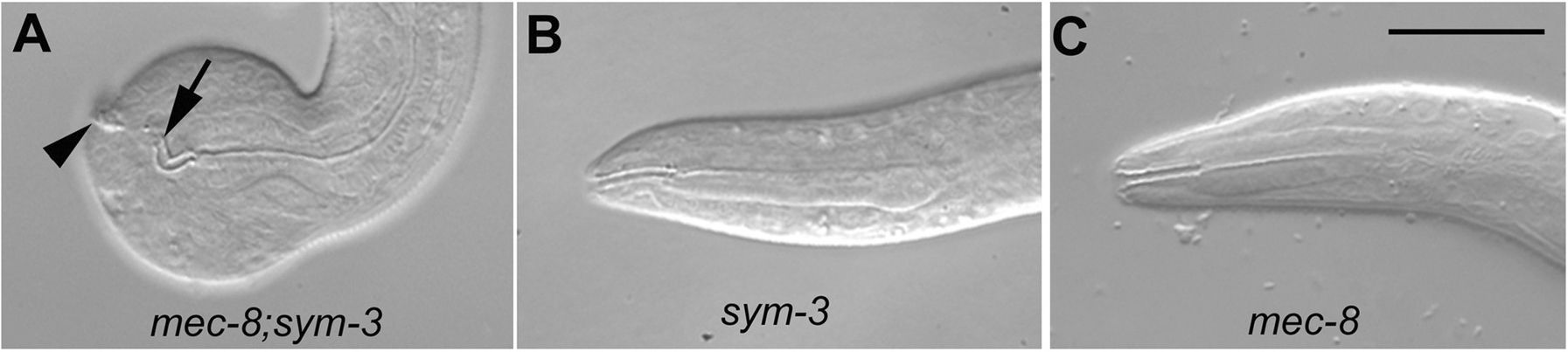
Figure 1. Synthetic interaction between mec-8 and sym-3 . (A) The mec-8; sym-3 double mutant has a highly penetrant defect: the anterior tip (arrow) of the pharynx is not properly joined to the anterior tip of the body (arrowhead), and a functional mouth is not formed. Unable to feed, hatchlings arrest growth and development during the first larval stage. The mutants also have a bulbous nose, an enlargement of the anterior-most part of the body, which is particularly evident in the example shown here. The junction of the pharynx and anterior end of the body is normal in the sym-3 single mutant (B) and in the mec-8 single mutant (C). Scale bar, 20 μm.
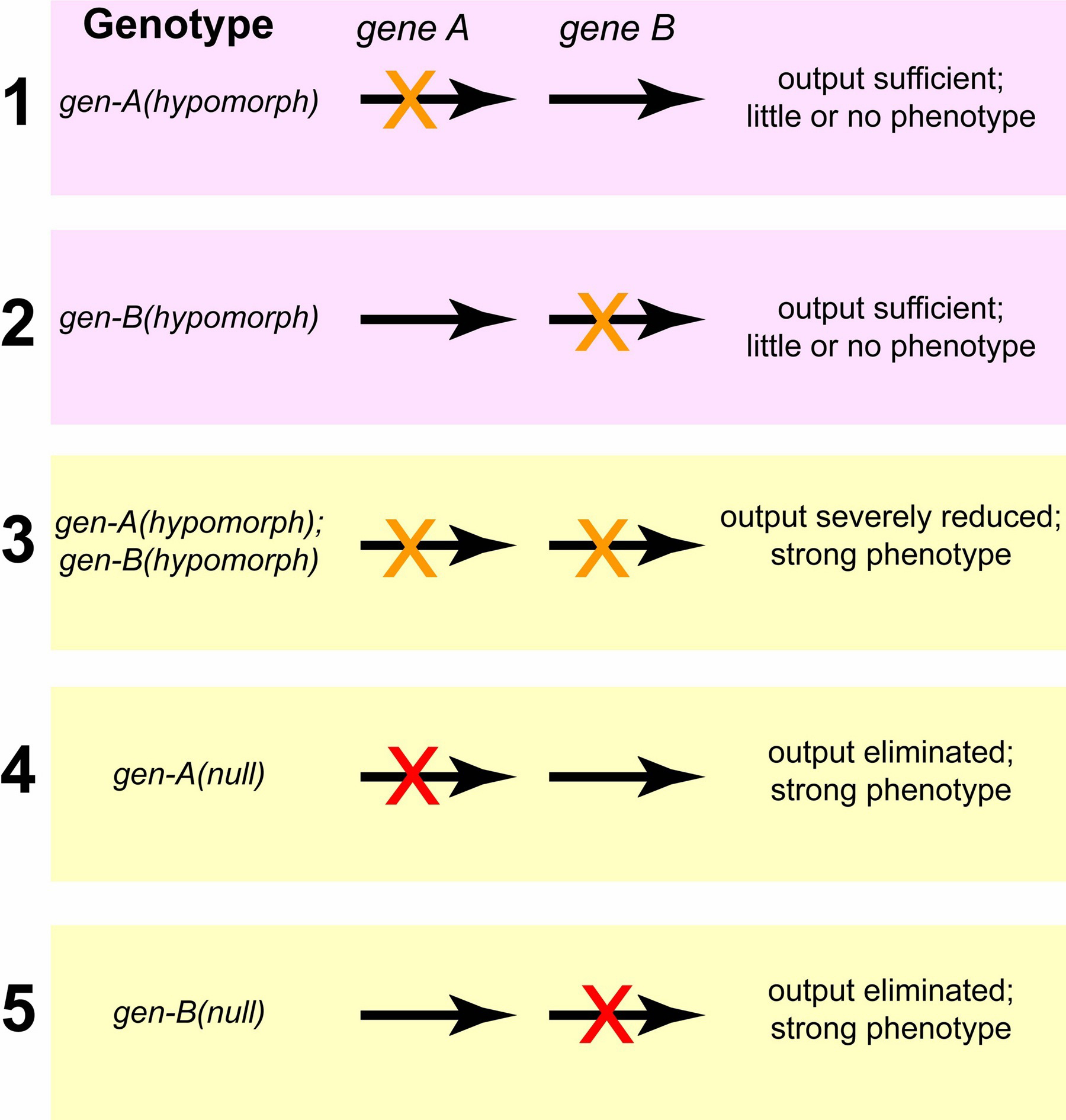
Hypomorphic mutations that enhance each other, by contrast, may be in genes that work in the same pathway; the two wild-type genes may act in the same direction, either positively or negatively, to regulate a downstream function, in which case the two hypomorphic mutations can synergize to give a phenotype stronger than the phenotype caused by either mutation on its own (Figure 2). We emphasize that two mutually-enhancing hypomorphic mutations need not always reflect involvement of the two genes in the same pathway or protein complex; impairment but not elimination of two redundant pathways by hypomorphic mutations might result in an insufficient level of final product or output.
Intergenic noncomplementation refers to the situation in which two mutations are in different genes and fail to complement despite being fully recessive to their wild-type alleles when present by themselves; i. e., each single heterozygote has a wild-type phenotype, but the double heterozygote exhibits a mutant phenotype. Each mutation can be thought of as a genetic enhancer of the other mutation. Intergenic noncomplementation is generally rare, and only a few examples are known in C. elegans, but they are instructive. Various alleles of the genes sqt-1, sqt-2, sqt-3, and rol-8 are recessive to their wild-type alleles on their own but show intergenic noncomplementation in certain combinations to give a twisted cuticle, which may be left-handed or right-handed, depending on the specific mutations involved (Kusch and Edgar, 1986). The alleles that behave in this way are not null, and it was proposed that they produce proteins that interact to disrupt the cuticular architecture. It is therefore not surprising that sqt-1, sqt-2, sqt-3 and rol-8 all encode cuticular collagens (Kramer et al., 1988; Cox et al., 1989; van der Keyl et al., 1994).
Mutations in genes that affect chemical synapse function have also provided good examples of intergenic noncomplementation (Yook et al., 2001; see Complementation). For example, certain recessive mutations in unc-13 and unc-64, which encode proteins required for the exocytosis step of the synaptic vesicle cycle, fail to complement. As in the cuticle examples, noncomplementation does not occur when both mutations are null, which means that noncomplementation is not simply the result of reduced dosage of both gene products but depends on the poisoning effect of a partially functional protein. Noncomplementation was also observed between mutations in other genes involved in synaptic vesicle trafficking, including genes whose products are thought not to interact directly. Allele-specific intergenic noncomplementation between mutations in genes that act in a pathway for male tail ray development has also been reported (Ikegami et al., 2004).
|
Figure 2. Enhancement between hypomorphic mutations in two genes that act sequentially in a pathway. A hypomorphic mutation in the first gene results in less product for the second step, which requires the second gene. If this gene is also crippled by a hypomorphic mutation, the final product or output of the pathway can be below a critical threshold. Hypomorphic mutations in two genes can also enhance each other if the genes encode components of a complex. A complex with only one crippled component may still have adequate function, but the presence of two crippled components, as occurs in the double mutant, may cause complete inactivity or disruption of the complex.
Dominant enhancement that does not involve intergenic noncomplementation has been seen in an analysis of animals homozygous for a temperature-sensitive mutation in one of several mec genes that affect the response to light touch (Gu et al., 1996). It was found that many otherwise-recessive mutations in other mec genes dominantly enhance the mechanosensory defects conferred by the temperature-sensitive mutations. The results were consistent with a model in which the interacting genes encode components of a protein complex that transduces mechanosensory signals. Finally, a myo-3 null mutation dominantly enhances the phenotype of a homozygous unc-54 mutant (Waterston, 1989); myo-3 and unc-54 encode different forms of myosin heavy chain, both present in body wall muscle. The enhancement in this case is due to reduced dosage of myo-3 product; conversely, enhanced expression of MYO-3 suppresses the uncoordinated phenotype conferred by the loss of UNC-54 (Riddle and Brenner, 1978; Maruyama et al., 1989).
Only 20-35% of all C. elegans genes when inactivated by mutation give a lethal, sterile, or obvious visible phenotype (Johnsen and Baillie, 1991; Waterston et al., 1997; C. elegans Sequencing Consortium, 1998; Hodgkin and Herman, 1998; see Essential genes). The many genes with nearly silent null alleles may contribute in subtle but significant ways to the fitness of the worm in nature, perhaps under particular environmental conditions, but the mild effects of knockout mutations make it difficult to study the specific roles of these genes. An obvious challenge is to determine what all these additional genes are doing. Many genes with subtle effects contribute redundantly to visible or essential functions (Thomas, 1993; Cooke et al., 1997). Redundancy is apparent when the simultaneous inactivation of two genes results in a strong phenotype not seen with either single gene knockout. In this case, each mutation is acting as an enhancer (usually recessive) of the other mutation. Complete gene redundancy, in which two genes perform exactly the same role and either can be inactivated with no loss of fitness, may be rare (Brookfield, 1997), but partial gene redundancy seems to be common, and many examples, again generally involving mutually-acting recessive enhancers, have been studied in C. elegans, and several are cited in the following two sections. Possible selective advantages of gene redundancy have been discussed (Thomas, 1993; Cooke et al., 1997); one idea, for example, is that partially redundant genes may together increase the fidelity of a biological process. Whatever the benefit of redundancy for the organism, there are obvious benefits to the researcher in studying redundant genes in appropriate double (or multiple) mutants.
Null or near-null mutations were identified in three genes that encode different kinetic classes of acetylcholinesterase (Culotti et al., 1981; Johnson et al., 1981; Johnson et al., 1988). Single mutants and ace-1 ace-3 and ace-2 ace-3 double mutants exhibit wild-type or nearly wild-type behavior, but ace-1 ace-2 mutations enhance each other, causing a severe uncoordination in the homozygous double mutant. Finally, the ace-3 mutation is an enhancer in the triple mutant, which is inviable—a synthetic lethality. Clearly, certain acetylcholinesterase functions are provided redundantly.
The genes lin-12 and glp-1 were originally identified on the basis of mutations that led to cell fate transformations. The genes were subsequently shown to encode similar Notch-like transmembrane receptors. The single mutant phenotypes are distinct, but the lin-12 glp-1 double mutant exhibits a much more severe phenotype than that of either single mutant (Lambie and Kimble, 1991). It was concluded that lin-12 and glp-1 provide some developmental roles individually, which are apparent in single mutants, but that they also provide some functions redundantly, which is made evident by the synthetic lethality of the double mutant.
Many examples of enhancement between mutations in homologous genes are now known, largely as a consequence of the availability of the genome sequence, which has given a complete picture of duplicated genes and gene families (see Gene duplications and genetic redundancy in C. elegans). Often, a mutation is identified that gives a phenotype (perhaps a subtle one); the gene is cloned; a homolog in the worm genome is found; and then either a knockout mutation of the second gene is sought, or RNA interference (RNAi) is used to reduce its expression; the phenotypes of single gene knockouts are then compared to the double knockout. Table 1 is an incomplete list of examples in which enhancement has been found by this general strategy. In all of these cases, it was concluded that the functions of the two members of each pair of genes overlap at least partly.
There are also cases of functional overlap involving three homologous genes, as was seen above for the ace genes. For example, the three worm rac-like genes function redundantly in axon pathfinding (Lundquist et al., 2001), as shown by enhancement of axon defects in various mutant combinations or mutants treated by RNAi; and a combination of RNAi and mutation in a sensitized genetic background has been used to demonstrate that the genes apx-1, dsl-1, and lag-2 encode redundantly-acting ligands for LIN-12 signaling during vulva development (Chen and Greenwald, 2004).
Overlapping functions can also be supplied by nonhomologous genes, which can be assigned to distinct but functionally overlapping pathways when animals carrying null mutations in both exhibit a more severe or more highly penetrant mutant phenotype than that found for either single mutant. In this way, genes affecting engulfment of cell corpses have been assigned to two parallel pathways (Ellis et al., 1991), as have genes that regulate dauer larvae formation (Malone and Thomas, 1994), sex myoblast migration (Chen et al., 1997), entry into meiosis (Kadyk and Kimble, 1998; Hansen et al., 2004), embryonic morphogenesis (Harrington et al., 2002), and ventral axon guidance (Hao et al., 2001).
Table 1. An incomplete list of homologous gene pairs exhibiting enhancement
| Gene pairs | Gene products or function | References | ||
|---|---|---|---|---|
|
Presenilins | Li and Greenwald, 1997 | ||
|
Regulatory myosin light chains | Rushforth et al., 1998 | ||
|
Drosophila nanos-like | Subramaniam and Seydoux, 1999 | ||
|
Transcription factors | Solari et al., 1999 | ||
|
Early embryonic patterning | Schubert et al., 2000 | ||
|
Medium chains of the AP-1 clathrin-associated protein complex | Shim et al., 2000 | ||
|
Regulate G-protein signaling | Dong et al., 2000 | ||
|
Zinc finger proteins | Detwiler et al., 2001 | ||
|
Semaphorins | Ginzburg et al., 2002 | ||
|
Modulate Ras-mediated processes | Ohmachi et al., 2002 | ||
|
Nonmuscle myosins | Piekny et al., 2003 | ||
|
Associate with P granules | Kawasaki et al., 2004 | ||
|
T-box transcription factors | Pocock et al., 2004 | ||
|
α-tubulins | Wright and Hunter, 2003; Phillips et al., 2004 | ||
|
β-tubulins | Wright and Hunter, 2003; Lu et al., 2004 |
Enhancers of null mutations are perhaps most instructive when they uncover surprising interactions. The classic worm example is provided by the synthetic multivulva (SynMuv) genes (Ferguson and Horvitz, 1989). The Muv phenotype (see Vulval development) requires two SynMuv mutations, one in a SynMuv class A gene and the other in a SynMuv class B gene. Two overlapping but nonhomologous pathways repress Muv development, which occurs only when both pathways are inactivated. Evidence for a third overlapping SynMuv pathway was recently published (Ceol and Horvitz, 2004). The original SynMuv interaction was discovered when it was found that a SynMuv mutant carried two unlinked mutations. Additional SynMuv genes were then found by screening for new SynMuv mutants in a class A or class B mutant background.
Screens for enhancers (usually recessive) of null mutations using extrachromosomal arrays provide a powerful method for discovering unanticipated overlapping functions. For example, mutations were sought that on their own conferred no obvious phenotype but which in combination with a null allele of mec-8, which encodes a putative RNA splicing factor, were synthetically lethal (Davies et al., 1999). Four genes identified in this way have been characterized (Davies et al., 1999; Yochem et al., 2004). Similarly, mutations have been identified that are synthetically lethal with a mutation in lin-35 (Fay et al., 2002), a class B SynMuv gene that encodes the worm homolog of the retinoblastoma protein. The interacting genes have been implicated in control of cell proliferation (Fay et al., 2002), pharyngeal morphogenesis (Fay et al., 2003; Fay et al., 2004), larval development (Cui et al., 2004), and somatic gonad development (Bender et al., 2004). Synthetic interactions with lin-35 and with other SynMuv genes in the control of cell-cycle progression have also been demonstrated by double-mutant and RNAi combinations (Boxem and van den Heuvel, 2001, 2002).
RNAi feeding of unc-34 null mutants has identified a synthetically lethal interaction between unc-34 and wve-1 (Withee et al., 2004); these two genes encode actin-regulating proteins that play overlapping roles in embryogenesis. In a genome-wide screen using RNAi feeding, gene interactions have been identified that are involved in the response to DNA damage (van Haaften et al., 2004). Genome-wide RNAi feeding is likely to become increasingly popular for identifying genetic enhancers, and an approach that uses extrachromosomal arrays is illustrated in Figure 3.
|
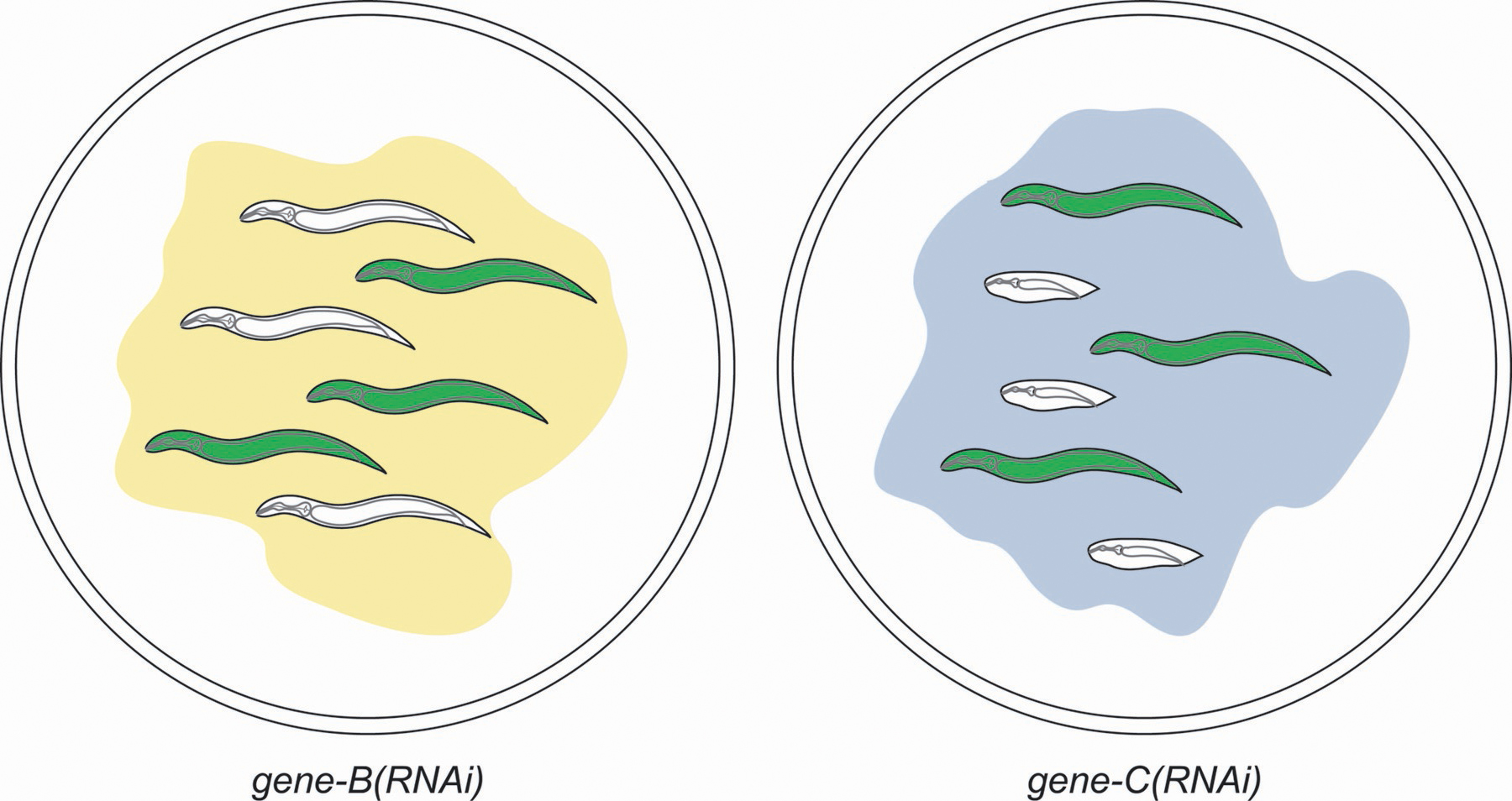
Figure 3. A screen for synthetic interactions based on RNAi-feeding libraries . The drawing depicts progeny of a mother that was chromosomally homozygous for a molecular null mutation in a gene (gene-A) that confers no obvious phenotype on its own. The mother was also transgenic for an extrachromosomal array that expresses a GFP marker and a wild-type version of gene-A. Some of the progeny have inherited the array and are gene-A(+) and green fluorescent, and others have not inherited the array and are therefore minus for gene-A and for the GFP marker. Being homozygous for the mutation in gene-A has no apparent effect on growth, development, and fertility of worms grown on a bacterial lawn (left plate) that expresses dsRNA that depletes gene-B. In contrast, a possible synthetic interaction between gene-A and gene-C is indicated by differential morphologies between array-plus (green) and array-minus (white) progeny when grown on a lawn of bacteria expressing dsRNA against gene-C (right plate). The lack of an effect on the transgenic [and therefore gene-A(+)] progeny indicates—but does not prove—that both genes A and C must be depleted before a phenotype is manifested.
Screens for recessive enhancers of partial loss-of-function mutations are often designed to identify additional components acting in the same pathway. For example, in a screen for enhancers of a weak glp-1 mutation affecting germline proliferation, novel alleles of two genes previously known to play essential roles in germline development were identified as well as mutations in five new genes (Qiao et al., 1995). One of the latter mutations was a weak allele of a gene called ego-1, which encodes a predicted RNA-directed RNA polymerase that functions both in germline development and in the process of RNA interference (Smardon et al., 2000). A screen for enhancers of defects in egg-laying and the excretory system caused by a hypomorphic lin-45 raf mutation yielded unusual alleles of several known Ras pathway genes, confirming that the screen worked as intended, and two new genes, eor-1 and eor-2 (Howard and Sundaram, 2002; Rocheleau et al., 2002). Apparent null alleles of eor-1 and eor-2 on their own (or in eor-1 eor-2 double mutants) cause only low penetrant defects, but they synergize with other Ras pathway mutations, suggesting that they play a modulatory role in Ras pathway signaling. Indeed, strong enhancement of null alleles of lin-25 and sur-2, which positively regulate Ras signaling, indicates that eor-1 and eor-2 provide a function that is functionally redundant with the function provided by lin-25 and sur-2 (Howard and Sundaram, 2002). In a screen for mutations that enhance a weak allele of mab-20, which encodes a semaphorin and plays a role in the formation of male tail rays, six genes were identified by enhancer mutations (Ikegami et al., 2004). One of these was the first known mutation in plx-2, which encodes a presumed receptor for MAB-20. Although mutations in plx-2 enhance a weak allele of mab-20, they do not enhance a null allele, indicating that plx-2 functions in the same pathway as mab-20. On the other hand, a putative null allele of plx-2, unlike mab-20 null, did not cause male ray defects on its own; genetic interactions between plx-2 and other genes argued for the existence of a parallel pathway in MAB-20 signal reception, involving an ephrin receptor.
Enhancer mutations can also be found among suppressor mutations (see Genetic suppression) when the suppressors are placed in a different genetic background. For example, suppressors of a lin-12 loss-of-function, not surprisingly, can enhance a lin-12 gain-of-function mutation (Sundaram and Greenwald, 1993). Conversely, sel-7 mutations, identified as suppressors of a lin-12 gain-of-function mutation, enhance a hypomorphic lin-12 mutation as well as a null allele of the presenilin gene sel-12 (Chen et al., 2004).
The phenotypic effects of some non-null alleles of unc-52 are greatly enhanced by loss-of-function mutations in mec-8 such that the mec-8; unc-52 double mutant exhibits the unc-52 null phenotype (Lundquist and Herman, 1994). The explanation for this result is that MEC-8 is required to promote specific alternatively-spliced mRNA isoforms that skip unc-52 exons that carry the enhanced mutations (Lundquist et al., 1996).
Enhancers have also been isolated in C. elegans that do not enhance the phenotype caused by a mutation in an endogenous worm gene; rather, they enhance a late-onset neuronal degeneration caused by an added transgene that expresses polyglutamine fragments (Faber et al., 2002). Recessive enhancer mutations in this case identified a gene that in its wild-type form can protect against polyglutamine toxicity. Thus, the examples of genetic enhancement in C. elegans are as multifarious as they are informative, and additional analyses should continue to enhance our knowledge of the genetic interactions within the worm genome.
Bender, A.M., Wells, O., and Fay, D.S. (2004). lin-35/Rb and xnp-1/ATR-X function redundantly to control somatic gonad development in C. elegans. Dev. Biol. 273, 335–349. Abstract Article
Boxem, M., and van den Heuvel, S. (2001). lin-35 Rb and cki-1 Cip/Kip cooperate in developmental regulation of G1 progression in C. elegans. Development 128, 4349–4359. Abstract
Boxem, M., and van den Heuvel, S. (2002). C. elegans class B synthetic multivulva genes act in G(1) regulation. Curr. Biol. 12, 906–911. Abstract Article
Brookfield, J.F. (1997). Genetic redundancy. Adv. Genet. 36, 137–155. Abstract
C. elegans Sequencing Consortium (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018. Article
Ceol, C.J., and Horvitz, H.R. (2004). A new class of C. elegans synMuv genes implicates a Tip60/NuA4-like HAT complex as a negative regulator of Ras signaling. Dev. Cell 6, 563–576. Abstract Article
Chen, E.B., Branda, C.S., and Stern, M.J. (1997). Genetic enhancers of sem-5 define components of the gonad-independent guidance mechanism controlling sex myoblast migration in Caenorhabditis elegans hermaphrodites. Dev. Biol. 182, 88–100. Abstract Article
Chen, J., Li, X., and Greenwald, I. (2004). sel-7, a positive regulator of lin-12 activity, encodes a novel nuclear protein in Caenorhabditis elegans. Genetics 166, 151–160. Abstract Article
Chen, N., and Greenwald, I. (2004). The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev. Cell 6, 183–192. Abstract Article
Cooke, J., Nowak, M.A., Boerlijst, M., and Maynard-Smith, J. (1997). Evolutionary origins and maintenance of redundant gene expression during metazoan development. Trends Genet. 13, 360–364. Abstract Article
Cox, G.N., Fields, C., Kramer, J.M., Rosenzweig, B., and Hirsh, D. (1989). Sequence comparisons of developmentally regulated collagen genes of Caenorhabditis elegans. Gene 76, 331–344. Abstract Article
Cui, M., Fay, D.S., and Han, M. (2004). lin-35/Rb cooperates with the SWI/SNF complex to control Caenorhabditis elegans larval development. Genetics 167, 1177–1185. Abstract Article
Culotti, J.G., Von Ehrenstein, G., Culotti, M.R., and Russell, R.L. (1981). A second class of acetylcholinesterase-deficient mutants of the nematode Caenorhabditis elegans. Genetics 97, 281–305. Abstract
Davies, A.G., Spike, C.A., Shaw, J.E., and Herman, R.K. (1999). Functional overlap between the mec-8 gene and five sym genes in Caenorhabditis elegans. Genetics 153, 117–134. Abstract
Detwiler, M.R., Reuben, M., Li, X., Rogers, E., and Lin, R. (2001). Two zinc finger proteins, OMA-1 and OMA-2, are redundantly required for oocyte maturation in C. elegans. Dev. Cell 1, 187–199. Abstract Article
Dong, M.Q., Chase, D., Patikoglou, G.A., and Koelle, M.R. (2000). Multiple RGS proteins alter neural G protein signaling to allow C. elegans to rapidly change behavior when fed. Genes Dev. 14, 2003–2014. Abstract
Ellis, R.E., Jacobson, D.M., and Horvitz, H.R. (1991). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics 129, 79–94. Abstract
Faber, P.W., Voisine, C., King, D.C., Bates, E.A., and Hart, A.C. (2002). Glutamine/proline-rich PQE-1 proteins protect Caenorhabditis elegans neurons from huntingtin polyglutamine neurotoxicity. Proc. Natl. Acad. Sci. USA 99, 17131–17136. Abstract Article
Fay, D.S., Keenan, S., and Han, M. (2002). fzr-1 and lin-35/Rb function redundantly to control cell proliferation in C. elegans as revealed by a nonbiased synthetic screen. Genes Dev. 16, 503–517. Abstract Article
Fay, D.S., Large, E., Han, M., and Darland, M. (2003). lin-35/Rb and ubc-18, an E2 ubiquitin-conjugating enzyme, function redundantly to control pharyngeal morphogenesis in C. elegans. Development 130, 3319–3330. Abstract Article
Fay, D.S., Qiu, X., Large, E., Smith, C.P., Mango, S., and Johanson, B.L. (2004). The coordinate regulation of pharyngeal development in C. elegans by lin-35/Rb, pha-1, and ubc-18. Dev. Biol. 271, 11–25. Abstract Article
Ferguson, E.L., and Horvitz, H.R. (1989). The multivulva phenotype of certain Caenorhabditis elegans mutants results from defects in two functionally redundant pathways. Genetics 123, 109–121. Abstract
Ginzburg, V.E., Roy, P.J., and Culotti, J.G. (2002). Semaphorin 1a and semaphorin 1b are required for correct epidermal cell positioning and adhesion during morphogenesis in C. elegans. Development 129, 2065–2078. Abstract
Gu, G., Caldwell, G.A., and Chalfie, M. (1996). Genetic interactions affecting touch sensitivity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 93, 6577–6582. Abstract Article
Hansen, D., Wilson-Berry, L., Dang, T., and Schedl, T. (2004). Control of the proliferation versus meiotic development decision in the C. elegans germline through regulation of GLD-1 protein accumulation. Development 131, 93–104. Abstract Article
Hao, J.C., Yu, T.W., Fujisawa, K., Culotti, J.G., Gengyo-Ando, K., Mitani, S., Moulder, G., Barstead, R., Tessier-Lavigne, M., and Bargmann, C.I. (2001). C. elegans slit acts in midline, dorsal-ventral, and anterior-posterior guidance via the SAX-3/Robo receptor. Neuron 32, 25–38. Abstract Article
Harrington, R.J., Gutch, M.J., Hengartner, M.O., Tonks, N.K., and Chisholm, A.D. (2002). The C. elegans LAR-like receptor tyrosine phosphatase PTP-3 and the VAB-1 Eph receptor tyrosine kinase have partly redundant functions in morphogenesis. Development 129, 2141–2153. Abstract
Hodgkin, J., and Herman, R.K. (1998). Changing styles in C. elegans genetics. Trends Genet. 14, 352–357. Abstract Article
Howard, R.M., and Sundaram, M.V. (2002). C. elegans EOR-1/PLZF and EOR-2 positively regulate Ras and Wnt signaling and function redundantly with LIN-25 and the SUR-2 Mediator component. Genes Dev. 16, 1815–1827. Abstract Article
Ikegami, R., Zheng, H., Ong, S.H., and Culotti, J. (2004). Integration of semaphorin-2A/MAB-20, ephrin-4, and UNC-129 TGF-β; signaling pathways regulates sorting of distinct sensory rays in C. elegans. Dev. Cell 6, 383–395. Abstract Article
Johnsen, R.C., and Baillie, D.L. (1991). Genetic analysis of a major segment [LGV(left)] of the genome of Caenorhabditis elegans. Genetics 129, 735–752. Abstract
Johnson, C.D., Duckett, J.G., Culotti, J.G., Herman, R.K., Meneely, P.M., and Russell, R.L. (1981). An acetylcholinesterase-deficient mutant of the nematode Caenorhabditis elegans. Genetics 97, 261–279. Abstract
Johnson, C.D., Rand, J.B., Herman, R.K., Stern, B.D., and Russell, R.L. (1988). The acetylcholinesterase genes of C. elegans: identification of a third gene (ace-3) and mosaic mapping of a synthetic lethal phenotype. Neuron 1, 165–173. Abstract Article
Kadyk, L.C., and Kimble, J. (1998). Genetic regulation of entry into meiosis in Caenorhabditis elegans. Development 125, 1803–1813. Abstract
Kawasaki, I., Amiri, A., Fan, Y., Meyer, N., Dunkelbarger, S., Motohashi, T., Karashima, T., Bossinger, O., and Strome, S. (2004). The PGL family proteins associate with germ granules and function redundantly in Caenorhabditis elegans germline development. Genetics 167, 645–661. Abstract Article
Kramer, J.M., Johnson, J.J., Edgar, R.S., Basch, C., and Roberts, S. (1988). The sqt-1 gene of C. elegans encodes a collagen critical for organismal morphogenesis. Cell 55, 555–565. Abstract Article
Kusch, M., and Edgar, R.S. (1986). Genetic studies of unusual loci that affect body shape of the nematode Caenorhabditis elegans and may code for cuticle structural proteins. Genetics 113, 621–639. Abstract
Lambie, E.J., and Kimble, J. (1991). Two homologous regulatory genes, lin-12 and glp-1, have overlapping functions. Development 112, 231–240. Abstract
Li, X., and Greenwald, I. (1997). HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc. Natl. Acad. Sci. USA 94, 12204–12209. Abstract Article
Lu, C., Srayko, M., and Mains, P.E. (2004). The Caenorhabditis elegans microtubule-severing complex MEI-1/MEI-2 katanin interacts differently with two superficially redundant β-tubulin isotypes. Mol. Biol. Cell 15, 142–150. Abstract Article
Lundquist, E.A., and Herman, R.K. (1994). The mec-8 gene of Caenorhabditis elegans affects muscle and sensory neuron function and interacts with three other genes: unc-52, smu-1 and smu-2. Genetics 138, 83–101. Abstract
Lundquist, E.A., Herman, R.K., Rogalski, T.M., Mullen, G.P., Moerman, D.G., and Shaw, J.E. (1996). The mec-8 gene of C. elegans encodes a protein with two RNA recognition motifs and regulates alternative splicing of unc-52 transcripts. Development 122, 1601–1610. Abstract
Lundquist, E.A., Reddien, P.W., Hartwieg, E., Horvitz, H.R., and Bargmann, C.I. (2001). Three C. elegans Rac proteins and several alternative Rac regulators control axon guidance, cell migration and apoptotic cell phagocytosis. Development 128, 4475–4488. Abstract
Malone, E.A., and Thomas, J.H. (1994). A screen for nonconditional dauer-constitutive mutations in Caenorhabditis elegans. Genetics 136, 879–886. Abstract
Maruyama, I.N., Miller, D.M., and Brenner, S. (1989). Myosin heavy chain gene amplification as a suppressor mutation in Caenorhabditis elegans. Mol. Gen. Genet. 219, 113–118. Abstract Article
Ohmachi, M., Rocheleau, C.E., Church, D., Lambie, E., Schedl, T., and Sundaram, M.V. (2002). C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr. Biol. 12, 427–433. Abstract Article
Phillips, J.B., Lyczak, R., Ellis, G.C., and Bowerman, B. (2004). Roles for two partially redundant α-tubulins during mitosis in early Caenorhabditis elegans embryos. Cell Motil. Cytoskeleton 58, 112–126. Abstract Article
Piekny, A.J., Johnson, J.L., Cham, G.D., and Mains, P.E. (2003). The Caenorhabditis elegans nonmuscle myosin genes nmy-1 and nmy-2 function as redundant components of the let-502/Rho-binding kinase and mel-11/myosin phosphatase pathway during embryonic morphogenesis. Development 130, 5695–5704. Abstract Article
Pocock, R., Ahringer, J., Mitsch, M., Maxwell, S., and Woollard, A. (2004). A regulatory network of T-box genes and the even-skipped homologue vab-7 controls patterning and morphogenesis in C. elegans. Development 131, 2373–2385. Abstract Article
Qiao, L., Lissemore, J.L., Shu, P., Smardon, A., Gelber, M.B., and Maine, E.M. (1995). Enhancers of glp-1, a gene required for cell-signaling in Caenorhabditis elegans, define a set of genes required for germline development. Genetics 141, 551–569. Abstract
Riddle, D.L., and Brenner, S. (1978). Indirect suppression in Caenorhabditis elegans. Genetics 89, 299–314. Abstract
Rocheleau, C.E., Howard, R.M., Goldman, A.P., Volk, M.L., Girard, L.J., and Sundaram, M.V. (2002). A lin-45 raf enhancer screen identifies eor-1, eor-2 and unusual alleles of Ras pathway genes in Caenorhabditis elegans. Genetics 161, 121–131. Abstract
Rushforth, A.M., White, C.C., and Anderson, P. (1998). Functions of the Caenorhabditis elegans regulatory myosin light chain genes mlc-1 and mlc-2. Genetics 150, 1067–1077. Abstract
Schubert, C.M., Lin, R., de Vries, C.J., Plasterk, R.H., and Priess, J.R. (2000). MEX-5 and MEX-6 function to establish soma/germline asymmetry in early C. elegans embryos. Mol. Cell 5, 671–682. Abstract Article
Shim, J., Sternberg, P.W., and Lee, J. (2000). Distinct and redundant functions of μ1 medium chains of the AP-1 clathrin-associated protein complex in the nematode Caenorhabditis elegans. Mol. Biol. Cell 11, 2743–2756. Abstract
Smardon, A., Spoerke, J.M., Stacey, S.C., Klein, M.E., Mackin, N., and Maine, E.M. (2000). EGO-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in C. elegans. Curr. Biol. 10, 169–178. Abstract Article
Solari, F., Bateman, A., and Ahringer, J. (1999). The Caenorhabditis elegans genes egl-27 and egr-1 are similar to MTA1, a member of a chromatin regulatory complex, and are redundantly required for embryonic patterning. Development 126, 2483–2494. Abstract
Subramaniam, K., and Seydoux, G. (1999). nos-1 and nos-2, two genes related to Drosophila nanos, regulate primordial germ cell development and survival in Caenorhabditis elegans. Development 126, 4861–4871. Abstract
Sundaram, M., and Greenwald, I. (1993). Suppressors of a lin-12 hypomorph define genes that interact with both lin-12 and glp-1 in Caenorhabditis elegans. Genetics 135, 765–783. Abstract
van der Keyl, H., Kim, H., Espey, R., Oke, C.V., and Edwards, M.K. (1994). Caenorhabditis elegans sqt-3 mutants have mutations in the col-1 collagen gene. Dev. Dyn. 201, 86–94. Abstract
van Haaften, G., Vastenhouw, N.L., Nollen, E.A., Plasterk, R.H., and Tijsterman, M. (2004). Gene interactions in the DNA damage-response pathway identified by genome-wide RNA-interference analysis of synthetic lethality. Proc. Natl. Acad. Sci. USA 101, 12992–12996. Abstract Article
Waterston, R.H. (1989). The minor myosin heavy chain, mhcA, of Caenorhabditis elegans is necessary for the initiation of thick filament assembly. EMBO J. 8, 3429–3436. Abstract
Waterston, R.H., Sulston, J.E., and Coulson, A.R. (1997). The genome. In: C. elegans II, D.L. Riddle, T. Blumenthal, B.J. Meyer, and J.R. Priess, eds. (Plainview, N.Y.: Cold Spring Harbor Laboratory), pp. 23–45. Abstract
Withee, J., Galligan, B., Hawkins, N., and Garriga, G. (2004). Caenorhabditis elegans WASP and Ena/VASP proteins play compensatory roles in morphogenesis and neuronal cell migration. Genetics 167, 1165–1176. Abstract Article
Wright, A.J., and Hunter, C.P. (2003). Mutations in a β-tubulin disrupt spindle orientation and microtubule dynamics in the early Caenorhabditis elegans embryo. Mol. Biol. Cell 14, 4512–4525. Abstract Article
Yochem, J., Bell, L.R., and Herman, R.K. (2004). The identities of sym-2, sym-3, and sym-4, three genes that are synthetically lethal with mec-8 in Caenorhabditis elegans. Genetics 168, 1293–1306. Abstract Article
Yook, K.J., Proulx, S.R., and Jorgensen, E.M. (2001). Rules of nonallelic noncomplementation at the synapse in Caenorhabditis elegans. Genetics 158, 209–220. Abstract
*Edited by Jonathan Hodgkin and Philip Anderson. Last revised March 14, 2005. Published September 16, 2005. This chapter should be cited as: Herman, R. K. and Yochem, J. Genetic enhancers (September 16, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.27.1, http://www.wormbook.org.
Copyright: © 2005 Robert K. Herman and John Yochem. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: bob-h@umn.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.