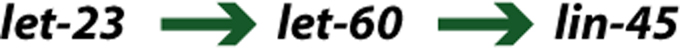Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
A major tool of developmental geneticists is the ordering of genes in functional pathways. In this chapter, we explain the logic behind constructing pathways, starting from the knowledge of the relevant phenotypes associated with the genes of interest, assuming that careful analysis of the phenotype has been carried out. We discuss the construction and interpretation of phenotypes of double mutants, screening for and analysis of extragenic suppressors, as well as issues regarding complex pathways and genetic redundancy. Avery and Wasserman (1992) have provided a brief theoretical discussion of epistasis analysis; here we explain the more practical aspects of how models of developmental pathways are built in C. elegans.
Epistasis is the masking of the phenotype of one mutant by the phenotype of a mutant in another locus. Hence, epistasis analysis can be used to determine a functional order of action of two genes, regardless of the directness of the interaction. However, epistasis analysis is most informative when the genes analyzed control a common process. Thus, it is important to determine the relationships between the mutations of interest before embarking on the construction of a formal genetic pathway.
Two different kinds of pathways exist (Figure 1). Historically, these have been distinguished as regulatory pathways and assembly or metabolic pathways. As both types of these pathways require "regulation," for the purpose of this chapter we will refer to the classical regulatory pathway as a switch regulation pathway and the classical assembly pathway as a substrate dependent pathway. The switch regulation pathway involves genes or gene products that can be turned "on" or "off". The different states of the genes (or their products) involved this type of pathway determine the outcome of the pathway. Mutations in genes involved in this type of pathway will have two distinct and opposite phenotypes, and will also have the ability to bypass the requirement for upstream genes. The second type of pathway involves a substrate, where an obligate series of sequential steps are required to generate the final outcome; classical examples of this second type of pathway include metabolic pathways and bacteriophage morphogenesis. Mutations in genes involved in the substrate dependent pathway will have phenotypes that suggest a progression of events. Sometimes determining which of the two types of pathways the genes of interest are involved in requires extensive phenotypic analysis of the mutations involved.
Consider for example, vulval differentiation in C. elegans (see Vulval development). In wild-type animals, three of six equipotential vulval precursor cells differentiate to form the vulva. These six precursor cells are a subset of the 12 "Pn.p" cells (the posterior daughters of the twelve ectoblasts, P1 through P12). Although loss-of-function mutants in lin-26 (lin= lineage abnormal), lin-39, and let-23 (let = lethal) are all vulvaless and have the same phenotype when examined under a dissecting microscope, these genes are involved in very different aspects of vulval differentiation. lin-26 is involved in the formation of the Pn.p cells, lin-39 is involved in determining which of the Pn.p cells become potential vulval precursor cells, and let-23 is involved in the fate decision regarding which Pn.p cells will actually differentiate into vulval tissue. Phenotypic analysis under higher magnification, Nomarski optics demonstrates that the vulvaless phenotype of animals mutant in lin-26 is due to the absence of Pn.p cells; in lin-39 mutants, the vulval precursor cells are not generated; and in let-23 mutants, the vulval precursor cells are not differentiating. In this example, the Pn.p cells are acting as the substrate while the genes lin-26, lin-39, and let-23 act to change the state of this cell. A mutation in each of these genes represents a step in the pathway that provides the appropriate context or substrate for the next step. Here, arrows are used to show a progression of events from one step to the next (Figure 1A). Epistasis analysis of substrate dependent pathways will confirm the order of events seen by phenotypic analysis.
On the other hand, mutations involved in a switch regulation pathway should all have the same phenotype or the direct opposite phenotype (Figure 1B). In effect, the mutations will represent two states of an event, either ON or OFF. An example of this type of pathway in C. elegans involves the tra-1 (tra= transforming) and her-1 (her= hermaphroditization) genes, which are needed for proper sex determination (see Somatic sex determination); these genes are part of a negative regulatory pathway and thus bars are used instead of arrowheads to symbolize the relationship between them. In C. elegans, the sex of the animals is determined by the number of X chromosomes in an animal; an XX animal is hermaphroditic while an XO animal is male. The her-1 loss-of-function mutation causes feminization of XO animals without affecting XX animals. The tra-1 loss-of-function mutation causes the opposite phenotype, masculinization of XX animals without affecting XO animals. These genes are involved in a switch regulation pathway because XO animals can be either one of two states, male or hermaphrodite; the same is true for XX animals.
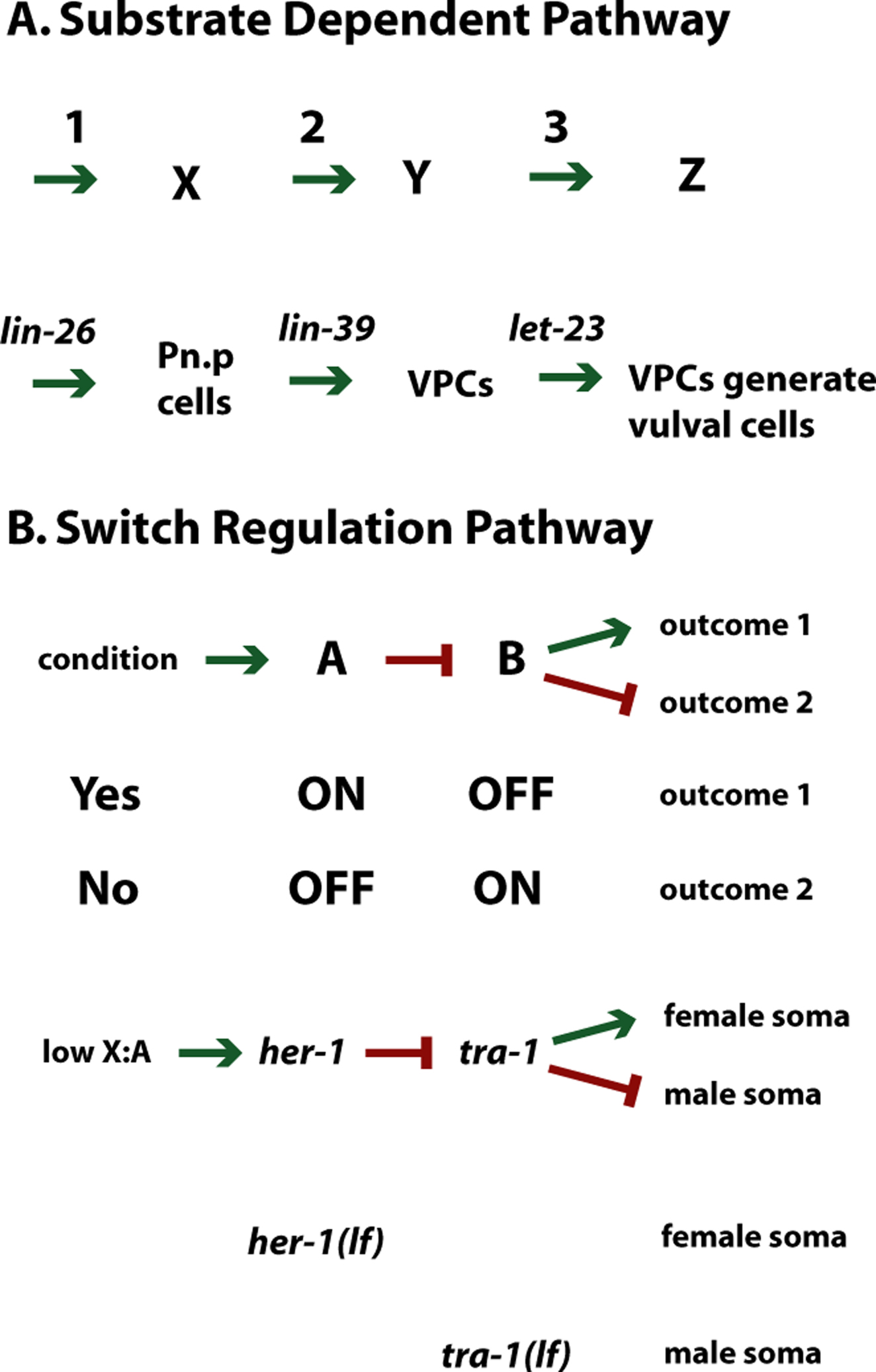 |
Figure 1. General types of pathways. (A) Substrate Dependent pathway: vulval development. A series of genes (1, 2 and 3) are necessary to produce a series of outcomes (X, Y and Z). lin-26 is necessary for the development of Pn.p cells. lin-39 is necessary to select a subset of Pn.p cells to become vulval precursor cells (VPCs). let-23 is necessary for the VPCs to generate vulval cells.(B) Switch Regulation pathway: somatic sex determination. A formal regulatory pathway is shown with conditions regulating which of two outcomes (1 or 2) will occur. If the condition is met (e.g., presence of a signal, particular environmental conditions; chromosome constitution), outcome 1 occurs. A specific example is shown at the bottom. High ratio of X chromosomes to autosome sets results in the activation of her-1 via a series of other genes. her-1 activity inactivates tra-1 via a series of other genes. tra-1 activity results in female somatic differention; tra-1 inactivity leads to male somatic differentiation. lf, loss-of-function mutation; arrows represent positive regulation; bars represent negative regulation.
Whether a gene should be placed in the definition of a switch regulation pathway or a substrate dependent pathway depends on the phenotype studied or the event assayed. In the study of the pathway containing the receptor tyrosine kinase encoded by let-23, its effects on the decision of the vulval precursor cells to take on vulval versus non-vulval fates constitutes a switch regulatory pathway. However, synthesis of the LET-23 receptor would constitute a substrate dependent pathway; one event would lead to another and the presence of LET-23 is required. Biochemical experiments do not necessarily define a substrate dependent pathway. Studies on how ligand activation of LET-23 leads to a phosphorylation cascade of downstream targets would be a switch regulation pathway; two distinct states are being analyzed, for example, phosphorylated and non-phosphorylated. Further, an obligate substrate is not needed in this pathway, as activation of a downstream target can bypass the need for LET-23 (e.g., a gain-of-function mutation in the let-60 ras gene will activate the pathway even in the absence of LET-23). It is imperative to distinguish whether the mutations of interest affect a switch regulation versus a substrate dependent pathway, as the logic used to order the genes involved is different and thus the resultant pathway constructed can be very different depends on a correct assessment. The ordering of genes in a substrate dependent pathway can be determined through an examination of the state of the substrate (i.e., the state of the Pn.p cell given above); epistasis analysis serves to confirm what is inferred from the phenotypic analysis or to imply the sequence of events. In the next section of this chapter, we will consider the logic behind constructing a switch regulation pathway, where pure phenotypic analysis will not allow gene ordering.
Once it is determined that the mutations of interest might constitute a switch regulation pathway, double mutants can be constructed to determine the epistatic relationships between the two mutations. The epistatic mutation is the one whose phenotype is displayed in the doubly mutant animal; the mutation whose phenotype is not displayed is hypostatic to the other. Thus, an epistasis test can only be performed on two mutations in different loci and with opposite phenotypes; epistatic relationships cannot be determined using mutations with the same phenotypes. However, genes with mutations of the same phenotype can be ordered by epistasis analysis using a mutation in a third gene that acts in the pathway between the two and that causes the opposite phenotype.
We outline three basic strategies for constructing double mutants in Figure 2. The first method is the simplest (Figure 2A) as it involves taking animals carrying the single mutations and using them directly to make the double mutants. Epistasis is determined by the self-progeny of the F2 animals. If animals of phenotype A produce progeny of phenotype A and B while animals of phenotype B only produce progeny of phenotype B, gene B is epistatic to gene A. Gene A would be epistatic to gene B if the opposite were true. Furthermore, the animals of phenotype B from parents of phenotype A will be the desired double mutant; these animals can be cultured to maintain the doubly mutant strain. Although this method is the simplest, it cannot be used if gene A and gene B are tightly linked on the same chromosome.
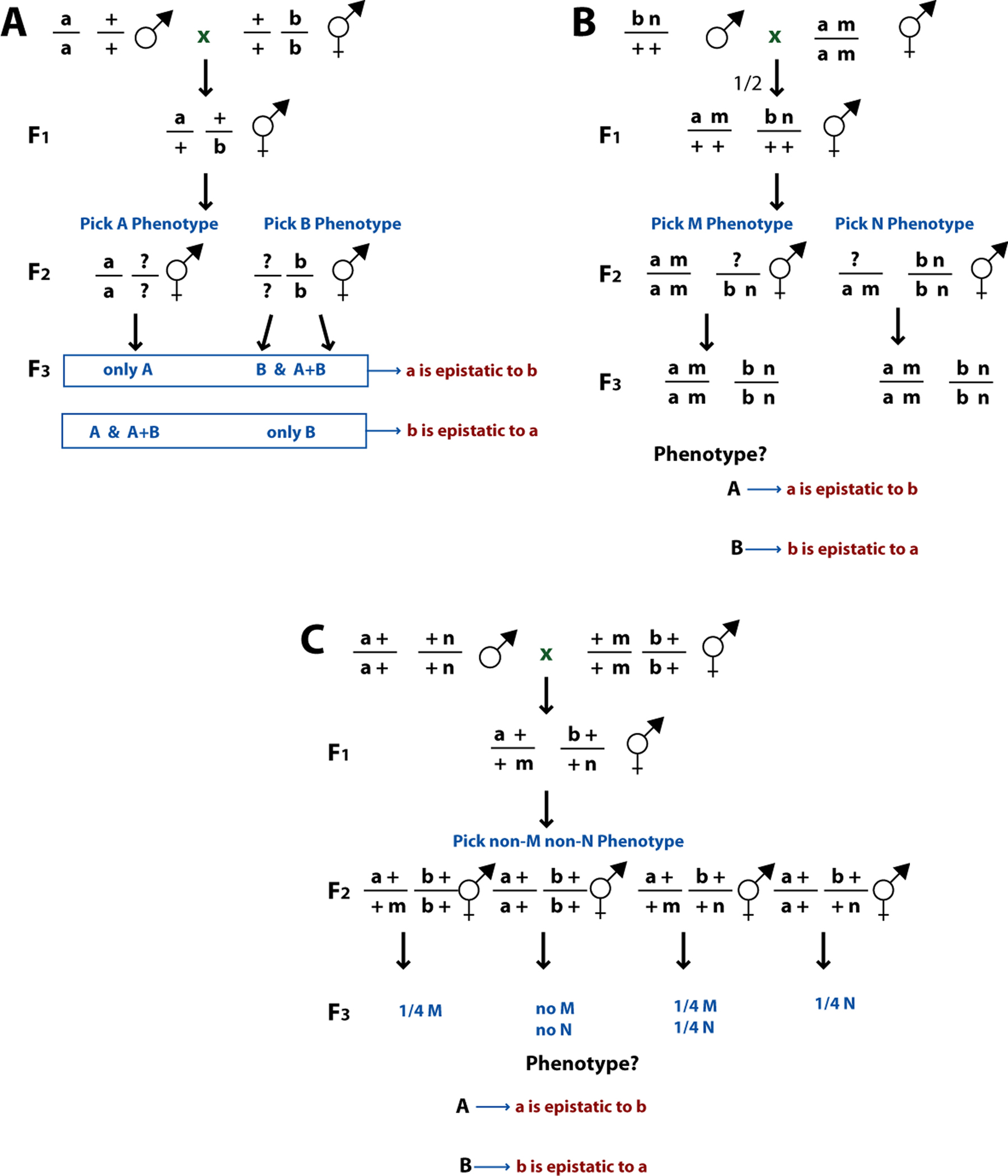 |
Figure 2. Construction of double mutant strains. (A) No markers. Animals carrying mutations in one gene of interest (gene A) with phenotype A are mated with animals carrying mutations in the other gene of interest (gene B) with phenotype B. The F1 heterozygote (gene a/+; gene b/+) will produce progeny of both phenotype A and phenotype B. Next, F2 animals of phenotype A and phenotype B are individually cultured and their F3 progeny examined to determine whether animals of phenotype A produce progeny of phenotype B or vice versa. By analyzing both classes, one can ensure that the desired strain will be constructed, even if one's assumption about the double mutant phenotype is incorrect. "?" indicates the allele at a given locus in a particular animal is not inferable from its phenotype or the genotype of its parents. (B) Linked markers in cis. The second method used for epistasis analysis involves using linked markers in cis to (on the same chromosome as) the genes of interest. The markers linked to the two genes should be different, closely linked to the gene of interest to avoid recombinants, not have any epistatic relationships to each other, not have any interactions with the genes of interest, and be easily scored as a double mutant. Males carrying mutation b with linked marker n are mated to hermaphrodites homozygous for mutation a and linked recessive marker m. Cross-progeny are non-M and heterozygous for a and b. Cross-progeny that do not produce N progeny are discarded, as they did not receive the b n chromosome from their father. From this heterozygote, hermaphrodites with phenotype M or N are picked, and the double mutant M N (also homozygous for a and b) is easily recognized. (C) Linked markers in trans. The best method for determining epistatic relationships involves using markers (m, n) linked in trans to (on the homologous chromosome as) the genes of interest. This will eliminate non-specific marker effects while ensuring that the double mutation of the two genes of interest is actually being constructed. Once again, the markers should be fairly close to the gene of interest to reduce the possibility of recombination. This construction is similar to that in part A, except that doubly mutant animals can be ascertained by the method described above and also by their failure to produce progeny carrying the markers linked in trans.
The second method used for construction involves using a closely linked marker in cis to (on the same chromosome as) each gene of interest (Figure 2B). The construction then involves making the strain carrying both marked chromosomes. The advantage of this method is that the strain can be constructed by simply following the mutations used as markers without regard to the phenotypes in interest. Once the doubly marked animal is obtained, the phenotype of interest can be determined. If the doubly marked animal has phenotype A, gene A is epistatic to gene B and vice versa. However, the control experiment must be done to construct double mutants with just the two markers, to make sure there is no effect on the phenotype of interest. Sometimes this method cannot be used because the markers will occlude scoring the phenotypes of interest.
The best method for determining epistatic relationships involves using linked markers in trans to (on the homologous chromosome as) the mutations of interest (Figure 2C). This will eliminate non-specific marker effects, while ensuring that the double mutation of the two genes of interest is actually being constructed. The markers should be close to the gene of interest to reduce the possibility of recombination. These markers can either be mutations in an unrelated gene or, better yet, balancer chromosomes (see Edgley et al. (1995) for an explanation of how to use balancers). This construction is similar to the first construction described, except that doubly mutant animals can be ascertained by the methods described above and also by their failure to segregate the trans markers.
A combination of methods can also be used. No matter which method is used to construct the double mutant, it is important to demonstrate that the presumptive doubly mutant animal actually carries both mutations of interest. One way that this can be done is by allowing males to mate with the doubly mutant hermaphrodites and picking F1 heterozygous animals. Animals of both phenotype A and phenotype B should be produced by the heterozygous animals, demonstrating that both mutations were present in the putative doubly mutant animal. Also, F1 males (which are presumably cross progeny and thus heterozygous) can be picked and outcrossed to test whether each male actually carries the A and B mutation.
In a switch regulation pathway, the epistatic gene is the downstream gene because these genes are negatively regulating each other. This rule can be derived from analysis of the single and double mutant phenotypes, as illustrated in the epistasis of two genes involved in sex determination, tra-1 and her-1. As mentioned above, the tra-1 mutation transforms XX animals into males while the her-1 mutation transforms XO animals into hermaphrodites. The tra-1; her-1 double mutant has the tra-1 phenotype; XX animals are males. tra-1 is epistatic to her-1 and thus downstream of her-1. tra-1 cannot be upstream of her-1 because the wild-type activity of tra-1 is to activate the hermaphrodite program of development while the wild-type activity of her-1 is to activate the male program of development. If tra-1 were upstream of her-1, inactivation of tra-1 would eliminate the repression of her-1, activating the male program of development. Inactivation of her-1 would lead to the inability to activate the male program of development, resulting in the hermaphrodite program of development. Removal of both tra-1 and her-1 would make tra-1 unable to repress her-1 (which does not matter because her-1 is inactivated by mutation) and her-1 unable to activate the male program of development. This would result in the double mutant exhibiting the her-1 phenotype. However, the experimental evidence says this is not true, so tra-1 is downstream of her-1; that is, inactivation of tra-1 obviates the need for her-1.
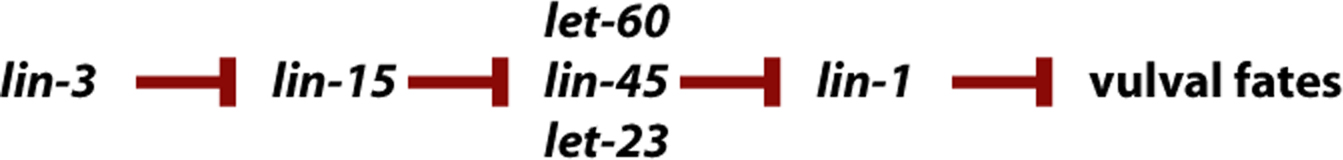
Comprehensive double mutant analysis with genes involved in a common process allows the construction of a pathway (see Figure 4). For example, during vulval induction, three of six vulval precursor cells (VPCs) normally adopt vulval fates while the other three adopt non-vulval hypodermal fates. By removing the inductive signal through ablation of the signaling cell, the three VPCs that normally become vulva now adopt non-vulval epidermal fates. Mutations that mimic the signaling-cell ablation phenotype (all six VPCs adopt non-vulval hypodermal fates, a.k.a. vulvaless (Vul) mutations) include let-23, lin-3, let-60, and lin-45 (see RTK/Ras/MAPK Signaling for details of the molecular nature). Mutations also exist that have the opposite phenotype (all six VPCs adopt vulval fates, a.k.a. multivulva (Muv) mutations); these include lin-15 and lin-1. Double mutation construction using Muv and Vul mutations yield the following results:
| lin-1 is epistatic to let-23 | let-23 is epistatic to lin-15 |
| lin-1 is epistatic to lin-3 | lin-15 is epistatic to lin-3 |
| lin-1 is epistatic to let-60 | let-60 is epistatic to lin-15 |
| lin-1 is epistatic to lin-45 | lin-45 is epistatic to lin-15 |
Since two states are being assayed, the ability of the VPCs to adopt vulval fates versus their ability to adopt non-vulval fates, the following pathway can be constructed, using the rule of the epistatic gene being the downstream gene (Figure 3):
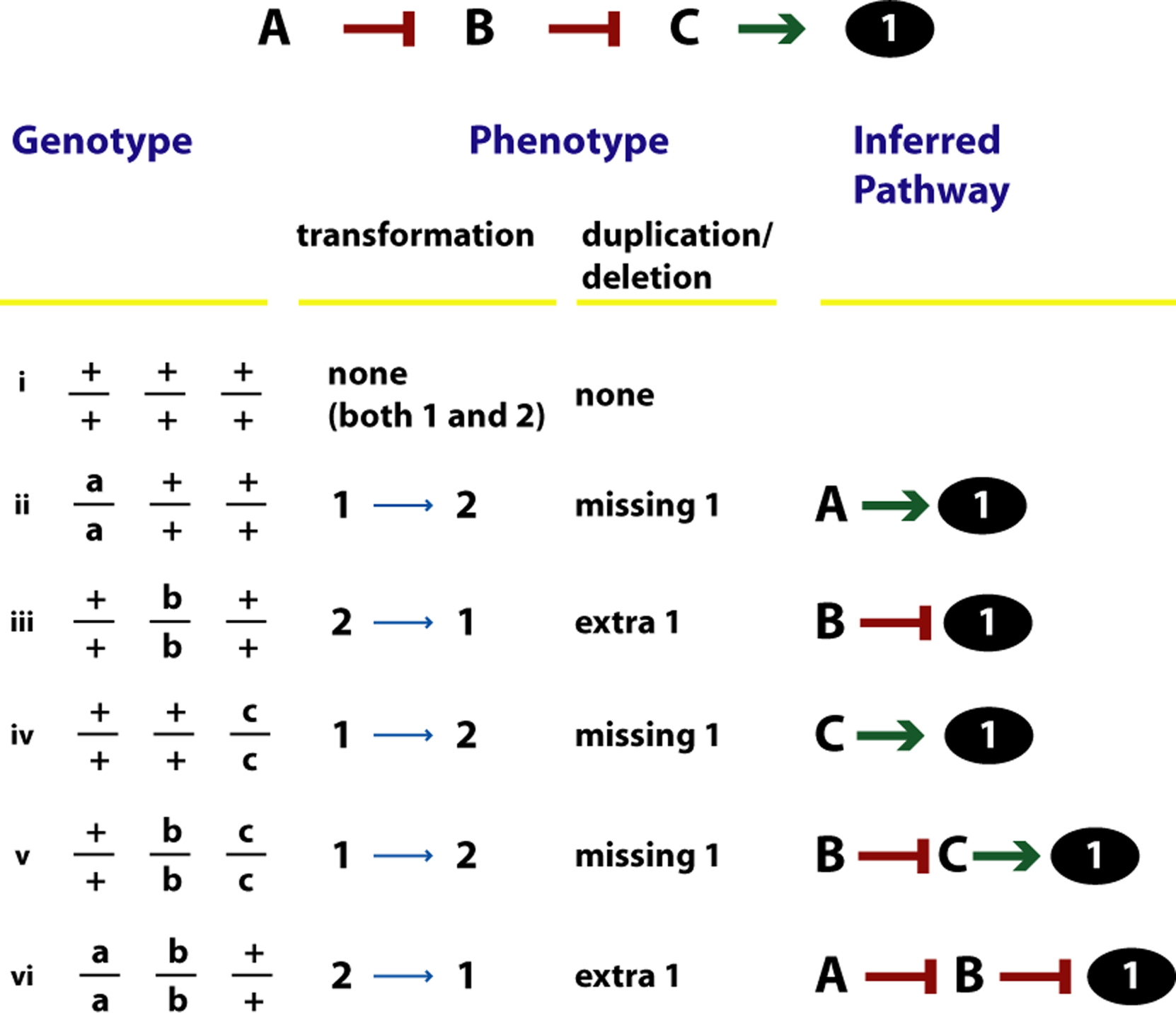 |
Figure 4. Epistasis analysis. The phenotypes of a set of genotypes and the inferences are shown. The inferred pathway leading to the outcome 1 (black oval; *) is shown at the top. The default outcome is 2. Phenotypes are described in two ways in this Figure: as transformations from 1 to 2 or 2 to 1, or as missing and extra structures, states or parameters. The inference from the phenotype of each genotype is shown. For example, in (ii), the absence of A leads to no 1; thus A promotes *. However, in (iii), the absence of B leads to extra 1; thus B inhibits *.
The examples of epistasis given above result from double mutant analyses using severe loss-of-function recessive alleles. Epistasis analysis is based upon the assumption that the mutations in the genes involved remove the function of that gene in the cell, tissue, or process being analyzed. Thus, it is important to determine by dosage analysis whether the alleles involved are hypermorphs (increased-function), hypomorphs (reduction-of-function), nulls (complete loss-of-function [also known as amorphs]) or neomorphs (novel function) [see Muller (1932) for details; see Sternberg (1990) and Greenwald & Horvitz (1980) for a discussion regarding the determination of a gene's null phenotype].
Null mutations are important for epistasis because the logic behind epistasis analysis is only valid if the two mutations used can be presumed to have no assayable activity. For example, consider two genes, A and B, in a regulatory pathway where there exists a null mutation in gene A but there exists no null mutations in gene B, only a hypomorphic mutation. If A is epistatic to B, this would not be as critical, since the normal function of B is to negatively regulate A. In this case, A is downstream of B. So, even if B has residual activity, the double mutant animals do not have functional A activity so the residual B activity cannot regulate A activity.
On the other hand, if B is downstream of A, the double mutant animal may display a misleading phenotype because the B mutation used contains residual activity and the doubly mutant animal has no A activity to regulate this residual activity. Since B and A have opposite phenotypes, having some wild-type B activity will lead to some expression of the A mutant phenotype, depending on how much activity the hypomorphic B mutation retains. Therefore, the outcome of an epistasis experiment where one or both of the mutations used are not null alleles could be co-expression, where both A and B phenotypes are expressed, or, a misleading double mutant phenotype, where the animal displays the A phenotype.
A practical example of this phenomenon can be found in Ferguson et al. (1987). Epistasis analysis using the lin-15 allele, n309, and the let-23 allele, n1045, resulted in the let-23(n1045); lin-15(n309) doubly mutant animals expressing either the Muv or the Vul phenotype. It is now known that let-23(n1045) makes some wild-type product (Aroian et al., 1994) and that lin-15(n309) is not a complete deletion of the locus (Huang et al., 1994; Clark et al., 1994). It is not possible to use a complete null allele of let-23 to analyze its phenotype in late larval development (without mosaic analysis) because, as the name implies, a let-23 null is lethal and would not survive to make the vulva. However, the sy97 allele of let-23 is a severe loss of function allele with respect to the vulva, such that vulval differentiation is essentially not seen (see Aroian and Sternberg, 1991 for an example of tissue-specific activities of a genetic locus). Molecular analysis also identified a lin-15 allele, e1763, whose lesion removes more coding sequence than n309. When epistasis analysis is performed using let-23(sy97) and lin-15(e1763), all the doubly mutant animals display a Vul phenotype (Huang et al., 1994). Of course, the let-23(sy97) allele did not exist in 1987 and the molecular nature of the lin-15 alleles was not known, so the epistasis was performed using the best of what existed and was known at the time.
As mentioned above, epistasis analysis cannot be performed using mutations that display the same phenotype. This can lead to subsets of the pathway where the order of some genes is not known. Sometimes these genes can be ordered through the use of gain-of-function alleles, which will display the opposite phenotype of the loss-of-function mutations. In these cases, it is important to know that the gain-of-function mutation in the gene of interest increases the normal activity of the gene and does not confer a novel activity.
In the example of the pathway regulating vulval differentiation given above, the genes let-60, let-23, and lin-45 cannot be separated by epistasis using hypomorphic alleles, as all display the Vul phenotype. However, there exists a gain-of-function allele of let-60; animals carrying this allele display the Muv phenotype. Double mutant construction using the let-60 gain-of-function allele and the lin-45 and let-23 hypomorphic alleles gives the following results:
| Genotype | Phenotype |
| lin-45; let-60(gf) | Vul |
| let-23; let-60(gf) | Muv |
Therefore, the let-60 gain-of-function allele can be used to split the order of action of let-23 and lin-45. These results imply the order (Figure 5):
Arrows are used because the wild-type function of the genes is to positively regulate each other. The gain-of-function phenotype of let-60 results in the opposite phenotype of hypomorphic let-60 alleles and allowed the ordering of these genes using the logic used for epistasis for switch regulation pathways.
Epistasis analysis does not always result in simple interpretations of gene order. Sometimes this is because the mutations used are not null alleles; the consequences of this are discussed above. Sometimes this is because the mutations used are not part of a switch regulation pathway but instead, part of a complex pathway that combines both switch regulation processes and substrate dependent processes. In fact, complex pathways are more often the rule than the exception. Development proceeds through a series of steps; often, a set of decisions about a fate leads to an execution of a particular fate, which may then lead to decisions about another fate. Development can be viewed as a set of substrate dependent pathways leading to switch regulation pathways which lead to more substrate dependent pathways, and so on.
Because the logic of epistasis differs for switch regulation and substrate dependent pathways, it is important to determine if the mutations involved in a complex pathway are all involved in the same step of the pathway being studied. This is not always obvious and may require careful phenotypic analysis. For example, animals carrying the mutation egl-18(n300) are Vul when examined under a dissecting microscope. Because animals carrying the lin-15 mutation are Muv, superficially, Muv and Vul are opposite phenotypes, the epistasis analysis of these two genes seems simple. The doubly mutant animal of genotype egl-18; lin-15 is Vul (Ferguson, et al., 1987); the simple interpretation is that egl-18 is downstream of lin-15. However, this is not the case. Careful examination of the egl-18 phenotype reveals its involvement in the generation of the VPCs; egl-18 animals are Vul because the VPCs are not there and thus cannot make a vulva. The correct interpretation is egl-18 is upstream of lin-15, operating in the pathway required to generate the VPCs and not in the regulation of VPC fate, as lin-15 does.
The process of programmed cell death is an excellent example of a complex pathway (see Programmed cell death; Figure 7). In C. elegans, 131 of the 1030 cells generated during development of the adult hermaphrodite undergo programmed cell death. Programmed cell death is a complex process, first involving the specification of whether or not a cell will die, followed by the process of killing the cell, and finishing with the engulfment and degradation of cell corpses. Mutations have been found in genes affecting each of these processes. For example, the genes ced-3 (ced=cell death abnormal), ced-4, and ced-9 are involved in the killing process (Ellis, et al., 1991b; Hengartner and Horvitz, 1992). Genes involved in the engulfment of cell corpses include ced-1, ced-2, ced-5, ced-6, ced-7, ced-8, and ced-10; mutations in these genes causes cell corpses to persist. The process of cell death thus consists of the steps outlined in Figure 6.
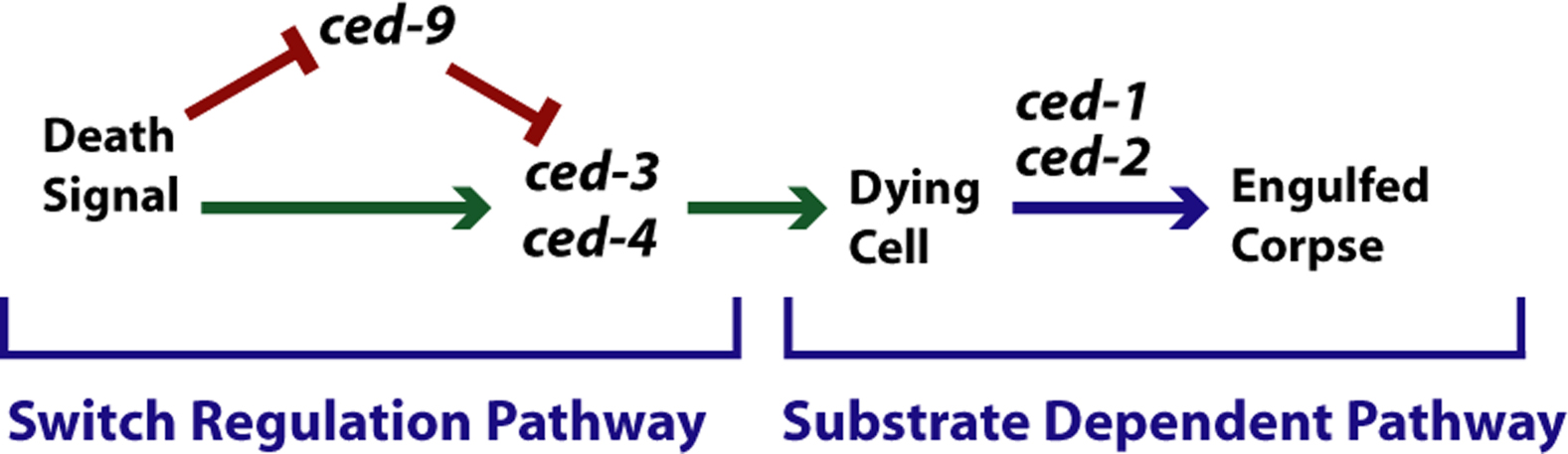 |
Figure 7. Mixed switch regulation and substrate dependent pathway: programmed cell death. An example of a mixed pathway. Some signal controls activities of either ced-9 or ced-3 and ced-4, resulting in activation of a pathway by which cells die. Inactivation of ced-9 results in extra cells dying. Inactivation of either ced-3 or ced-4 results in the survival of cells that would otherwise die. Inactivation of ced-1, ced-2, etc. results in cells that die, but whose corpses are not engulfed by surrounding tissue.
Epistasis can only be interpreted using the logic discussed for switch regulation pathways when the genes being analyzed are in a common process. In the example above, ced-3 and ced-1 are involved in different processes and thus phenotypic analysis will elucidate their order relative to each other such that this analysis will reveal the processes in which these genes are involved and thus reveal their order of action. In this case, an understanding of the events involved in cell death will demonstrate that cells are killed (which requires the action of ced-3) before they are engulfed (which requires the action of ced-1).
ced-3, ced-4, and ced-9 are involved in regulating the killing of cells. In animals without ced-3 or ced-4 function, cells survive that normally die by programmed cell death. Animals without ced-9 function die. When ced-9 mutant animals also lack either ced-3 or ced-4, they live, suggesting that functional ced-3 or ced-4 is necessary for the ced-9 gene product to function. This interpretation is consistent with the gain-of-function phenotype of ced-9, which results in the survival of cells that normally die by programmed cell death (this is the same phenotype as loss-of-function of ced-3 and ced-4). These results imply the gene order shown in Figure 8.
When double mutants are made using mutations in ced-3 and ced-1, a gene involved in the engulfment of cell corpses, the doubly mutant animal displays the ced-3 phenotype. However, ced-1 and ced-3 are part of the same substrate dependent pathway but not part of the same switch regulation pathway. For one, ced-3 and ced-1 do not have the opposite phenotype from one another. Phenotypic analysis would suggest that the cell must first be specified to die by ced-3 before its corpse can be engulfed through the activity of ced-1. Also, the engulfment pathway that ced-1 regulates is not necessary for causing cell death like ced-3, because mutations that prevent engulfment do not prevent most cell deaths (Hedgecock, et al, 1983; Ellis, et al, 1991a). The action of ced-3 leads to the action of ced-1. Thus, the pathway should be drawn as (Figure 9):
These examples regarding egl-18 and lin-15 in the vulval differentiation pathway and the genes in the programmed cell death pathway illustrate the importance of detailed phenotypic characterization of the singly mutant animals in preventing misinterpretation of double mutant phenotypes. Once it is determined that two genes are in a switch regulation pathway, the rule that the epistatic gene resides downstream of the hypostatic gene can be simply applied. In contrast, if two genes are involved in a substrate dependent pathway, double mutant analysis again confirms the order of events that have been visualized, although in this case, the epistatic gene is the upstream gene. Whether or not two genes are involved in a switch regulation process or a substrate dependent process can only be determined by careful analysis of the phenotypes of the mutations involved.
Another factor that can complicate the interpretation of epistatic relationships is genetic redundancy. Discussions on the theoretical implications of genetic redundancy can be found in Thomas (1993) and Tautz (1992); these issues are beyond the scope of this chapter. In practice, genetic redundancy can be discovered in three ways: by backcrossing newly isolated mutants of interest and discovering that the mutant phenotype is a result of two mutations, by constructing double mutants and discovering a new phenotype, or by undertaking a screen for redundant genes. These methods are discussed below (see Figure 10).
Discovering genetic redundancy by backcross involves examination of the frequency of segregation of the mutation involved from a heterozygous animal (Figure 10A). This method is most often used during the analysis of a new mutation following mutagenesis (for example, see Ferguson and Horvitz (1989)). A recessive phenotype caused by a single gene trait will usually segregate from a heterozygote animal at a frequency of 1/4. However, if two genes are required to cause the phenotype, animals expressing this phenotype will segregate from a heterozygous parent at a frequency of 1/16, assuming they are unlinked from each other. Once it is determined that two genes cause the phenotype of interest, the phenotype of each mutation alone should be determined. If these two mutations are genetically redundant, they will only cause the phenotype of interest in combination with each other and should display either a reduced phenotype alone or possibly even no phenotype.
Another method for discovering genetic redundancy involves constructing double mutant animals using existing mutations (Figure 10B). This is usually done when mutations are recovered that display an incomplete phenotype or that display no phenotype with respect to the phenotype of interest (i.e., a silent mutation). This method was extremely informative in the discovery of the two sets of genes involved in the engulfment process during programmed cell death (Ellis, et al., 1991a) and the redundancy seen in the dauer formation pathway (Thomas, et al., 1993).
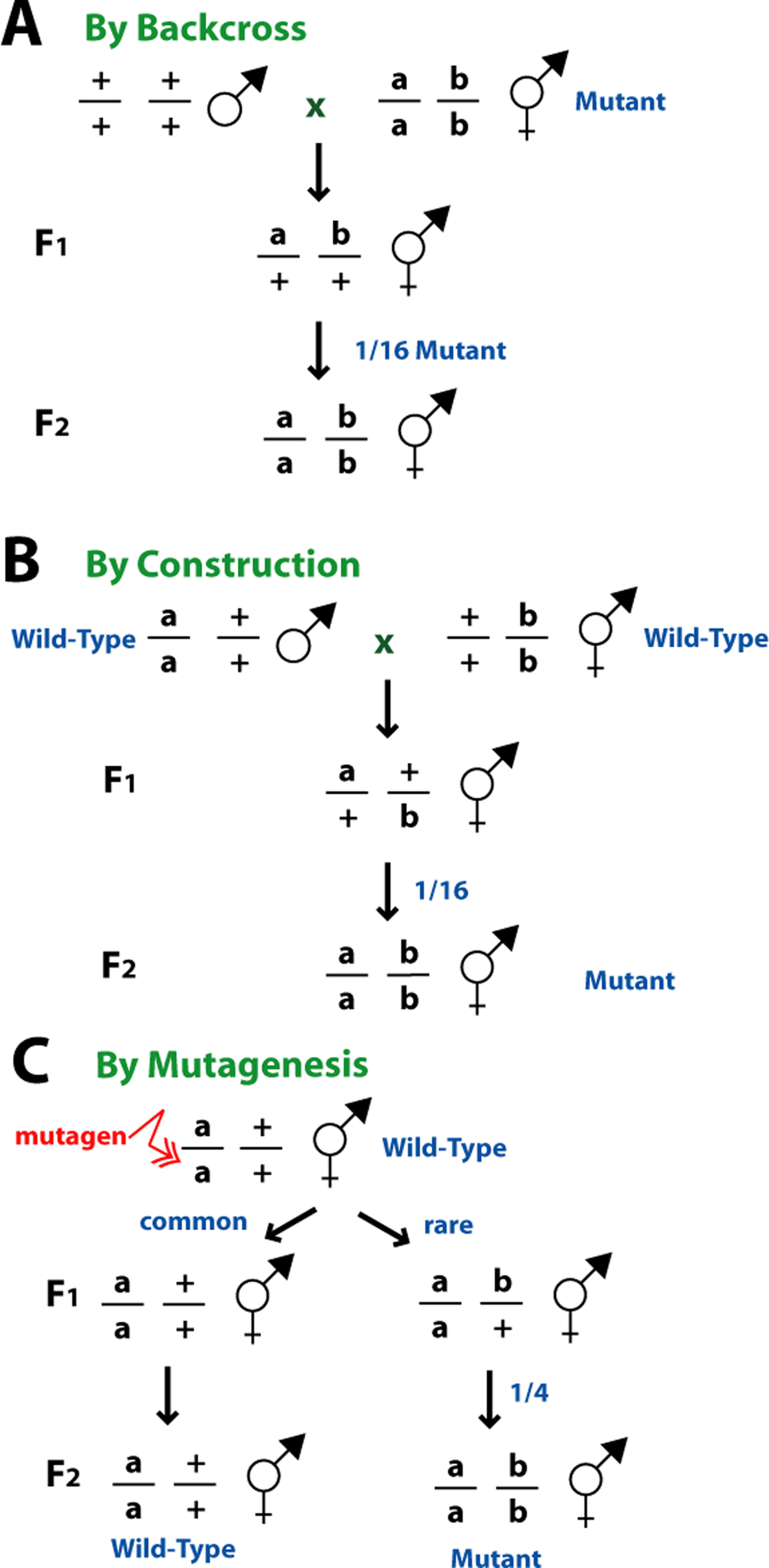 |
Figure 10. Synthetic phenotypes. (A) Discovery by backcrossing. If two recessive mutations are responsible for the phenotype, then 1/16 progeny of the double heterozygote will display the phenotype. (B) Discovery by construction. (C) Discovery by mutagenesis.
A third method for discovering genetic redundancy involves screening for new mutations that synergize with the original mutation to cause the phenotype of interest (Figure 10C, also see Ferguson and Horvitz, 1989). This is an especially useful technique if it is already known that redundancy exists in the system, through molecular information or by the discovery of genetic redundancy through other methods, as described above.
The formal interpretation of genetic redundancy is that the redundant genes are involved in parallel pathways, regulating the process of interest. For two redundant genes, A and B, that negatively regulate gene C that negatively regulates gene D, the pathway would look like (Figure 11):
In this case, the animal carrying mutations in both gene A and gene B would display the opposite phenotype as animals mutant in gene C and would display the same phenotype as animals mutant in gene D. Thus, when epistasis analysis is done with genetically redundant genes, both redundant genes should be eliminated because only then is the phenotype of interest seen. In this case, since gene A only partially negatively regulates gene C, even doing epistasis with a null allele of gene A will not completely eliminate the function of that particular step; both gene A and gene B function need to be completely eliminated to create the phenotype that represents a lack of function at that step. Thus, the triply mutant animal lacking gene A, gene B, and gene C must be constructed for epistasis analysis.
Although the ordering of genes in pathways using genetic epistasis is a powerful tool, it merely provides a working model for more phenotypic studies or molecular analysis and should not be taken as the “answer.” One limitation of epistasis is that it assumes a linearity of events. This is not always the case; as sometimes pathways are branched or contain multiple inputs. Also, some pathways use tissue specific regulators that are not necessarily used in other developmental processes that use a subset of these same genes. Ultimately, these issues can be resolved by careful phenotypic analysis of the genes involved in the pathway, although where to look may not be initially obvious.
One example of a branched pathway in C. elegans is the sex determination pathway (see Figure 12). A high X chromosome to autosome ratio (X:A) negatively regulates the fem-1, fem-2, and fem-3 genes, which negatively regulate the tra-1 gene. tra-1 functions to activate the developmental program for the female soma while repressing the developmental program for the male soma. However, the role of the fem-1, fem-2, and fem-3 gene products is not just to regulate tra-1; these genes are also responsible for promoting male germline development and negatively regulating female germline development. In the case of a high X:A ratio, inactive fem-1, fem-2, and fem-3 would lead to female germline development. By recognizing the branch point at fem-1, fem-2, and fem-3 and realizing that germline development is regulated differently than somatic development, the intersex phenotype of animals carrying the tra-1 mutation can be interpreted. XX tra-1 mutant animals have a male soma because they have no functional tra-1 to repress male somatic development; these XX animals also have a female germline because fem-1, fem-2, and fem-3 are repressed by the high X:A ratio.
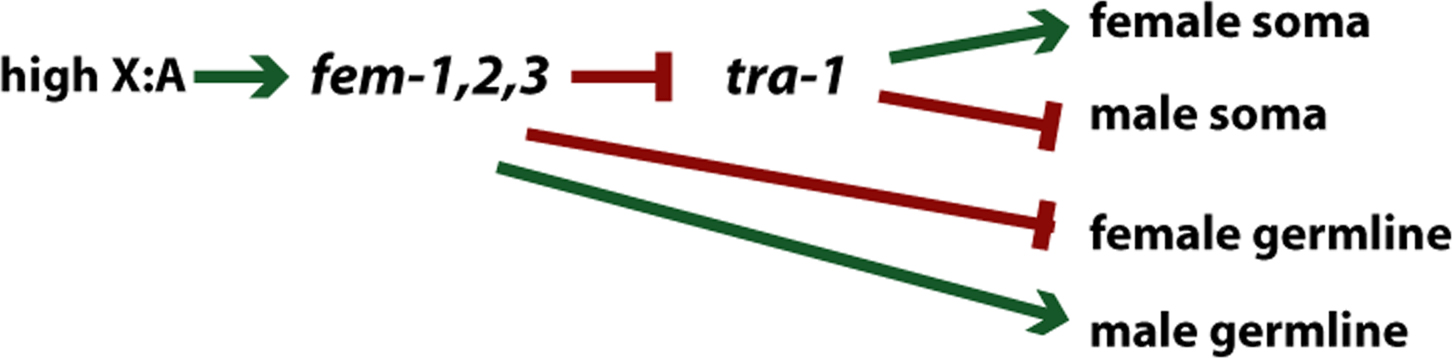 |
Figure 12. Branched pathway: somatic and germline sex determination. The ratio of X to autosomes determines sexual phenotype (see Villeneuve and Meyer (1990) for review). High ratio of X chromosomes to autosomes results in repression of activity of the fem genes. For somatic sexual determination, the fem genes negatively regulate tra-1 activity. For germline sex determination, the fem genes control sexual identity independently of tra-1.
This example also illustrates the existence of tissue specific regulators of a particular pathway. Although tra-1 is downstream of fem-1 in somatic development, it is not so in germline development. Thus, the order of gene action should be determined by epistasis analysis with the genes involved for the particular developmental process of interest, looking at their action in the tissue of interest. Another example of this is the use of the heterochronic genes lin-4, lin-14, lin-28, and lin-29 (Ambros and Moss, 1994). Animals carrying loss-of-function mutations in lin-4 and lin-14 have widespread defects in temporal control. By examining their effects in the temporal control of the hypodermal seam cells, a regulatory pathway was constructed with the more specifically used heterochronic genes, lin-28 and lin-29. A complete description of the construction of this pathway is found in Ambros (1989); this paper also contains an excellent explanation of how the logic of epistasis analysis was used to construct the pathway.
Another variation on branched pathways are pathways with more than one input (Figure 14). An example of this is the C. elegans vulval differentiation pathway. The pathway given in previous sections consists of (Figure 13):
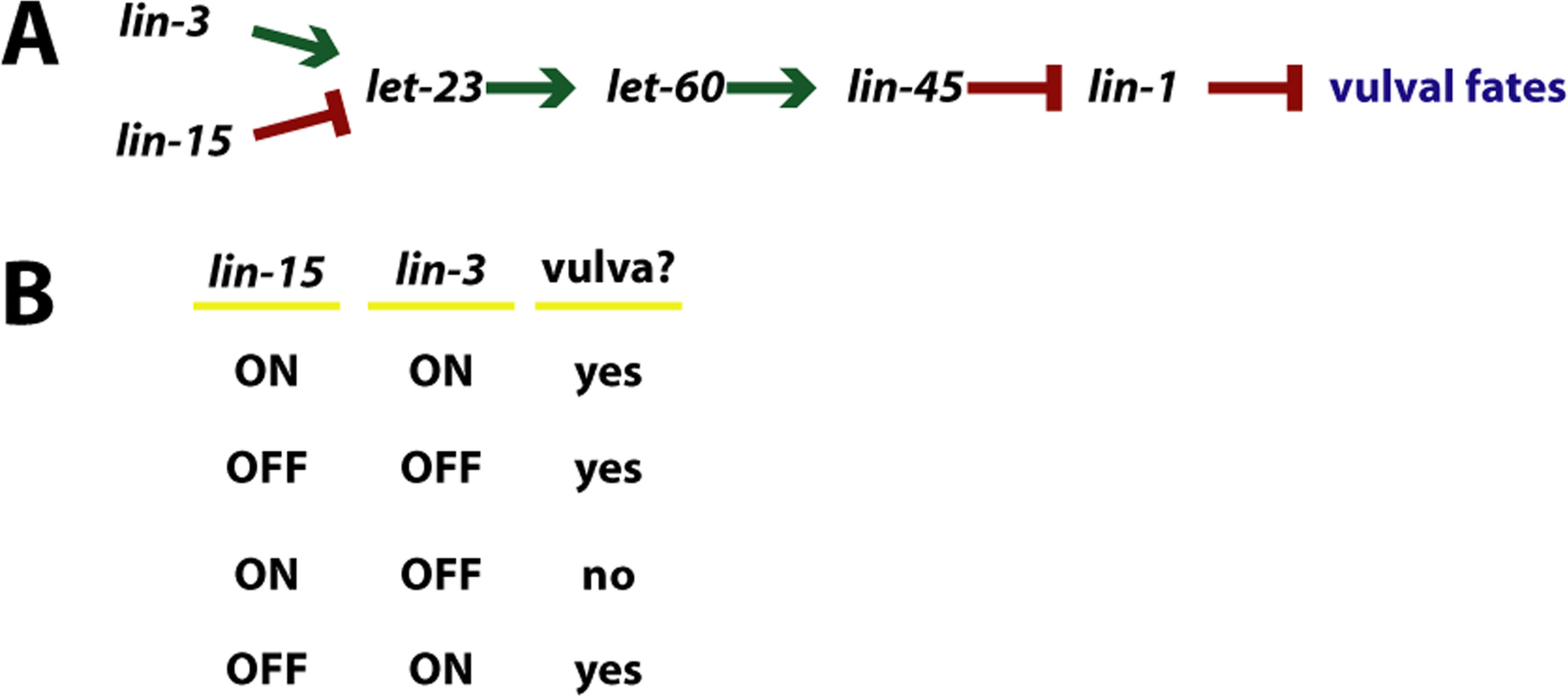 |
Figure 14. Multiple inputs: vulval induction. (A) Abbreviated pathway of vulval induction (see Sternberg (1993) for review). (B) A "truth table" schema for the combined action of lin-15 and lin-3 to control vulval differentiation.
However, the molecular identification of the lin-3 gene product as a growth factor/ligand-like molecule and the let-23 gene product as a receptor tyrosine kinase suggests that lin-3 encodes the ligand for let-23 (see RTKRas/MAP kinase signaling). Furthermore, previous phenotypic characterization of lin-15 demonstrated that although lin-15 mutant animals exhibit the mutant Muv phenotype even in the absence of the inductive tissue, their VPCs are still capable of responding to the signal. Thus lin-15 does not belong directly downstream of the lin-3 signal, but instead, as a second input upon let-23 (Figure 15):
The examples of the vulval differentiation pathway and the sex-determination pathway given above underscore the importance of careful phenotypic analysis in the construction of developmental regulatory pathways. This is not to say that epistasis analysis should not be performed until an exhaustive understanding of the mutant phenotype exists; however, pathways constructed through epistasis analysis should be considered models that can change over time, given new information from further phenotypic or molecular analysis.
The logic presented in this chapter can be extended to other non-genetic interactions, such as cell-cell interactions that can be dissected using cell ablations. Rather than remove the function of a gene by mutation, a cell can be removed by laser ablation (Avery and Bargmann, 1995) or by a pharmacological agent . One example of using cell ablations instead of mutation for epistasis is the analysis of cell signalling during male spicule development (Chamberlin and Sternberg, 1993). Bγ and Bδ are two different cell fates adopted by certain progeny of the B cell, a male specific blast cell involved in spicule development. Ablation of the nearby Y.p cell (the posterior daughter of the male specific blast cell, Y) leads to a defect in the δ lineage; ablation of the pa cells (the two cells, B.alpa and B.arpa, great-great granddaughters of the blast cell, B; sisters of γ and δ) leads to a defect in Bγ. As Bγ and Bδ are alternative cell fates, the ablation of the Y.p cell can be considered to cause the opposite phenotypes from that of the ablation of the pa cells. The double ablation of both Y.p and the pa cells has a defect similar to ablation of the Y.p cell alone. Using the logic of epistasis, the role of the pa cells might be to negatively regulate the effect of Y.p.
To further analyze a pathway, it is often desirable to identify additional genes. One method for doing this involves starting with a wild-type animal and screening for additional mutations with the phenotypes of interest. Another method involves isolating extragenic suppressor mutations of a previously identified mutant gene in the pathway (see Anderson (1995) for information regarding the different mutagens available). Suppression screens can be better than direct screens because the presence of the starting mutation might suppress the lethality of the suppressor mutation, and thus allow the mutation to be recovered. Screens for mutations that enhance a partial mutant phenotype can also identify new genes.
Screens for extragenic suppressors seem deceptively simple: a mutation in the pathway is chosen and a screen is performed to look for new mutations that no longer express the mutant phenotype, either in the F1 generation (for dominant suppressors) or in the F2 generation (for recessive suppressors) (see Figure 16). Although the screening process is straightforward, the key to the success of an extragenic suppressor screen involves choosing the most appropriate starting mutation for the purpose of the screen, as the choice of a starting strain affects the suppressor mutations isolated.
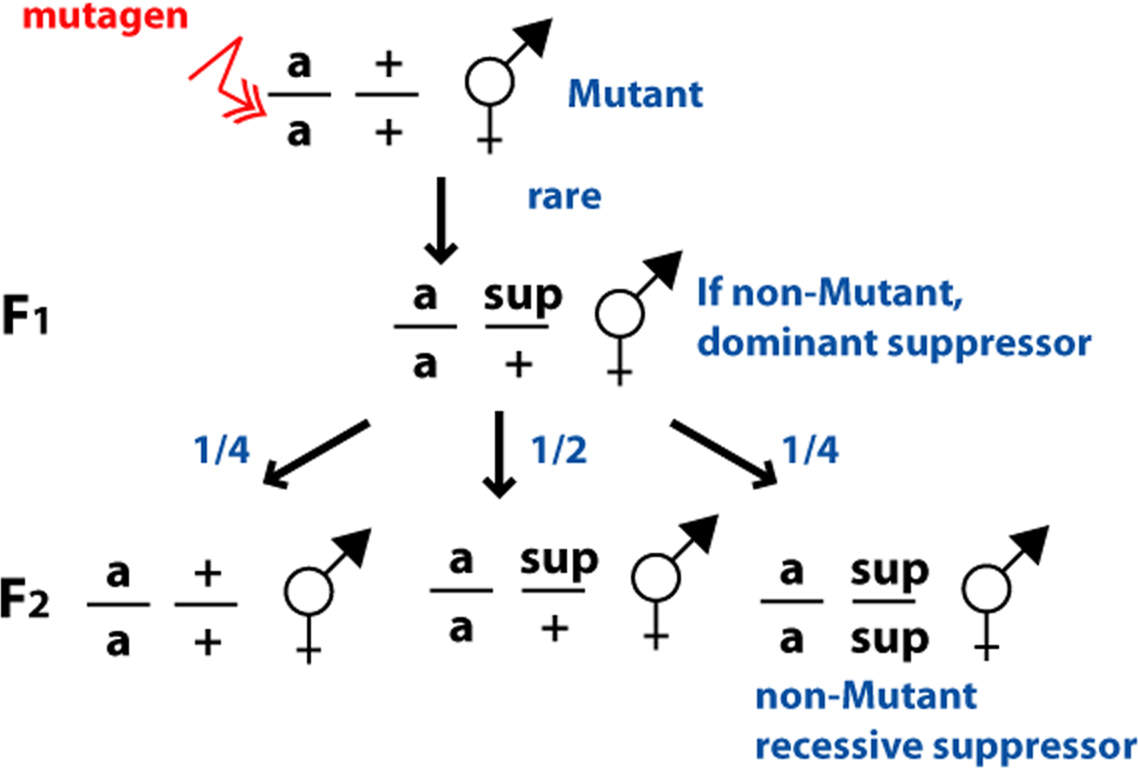 |
Figure 16. Suppressor screens. From mutant parents that have been mutagenized, the F1 progeny and F2 grandprogeny are examined for phenotypic revertants, that is animals that do not display the mutant phenotype. Each F1 examined screens two mutagenized gametes, as either the sperm or the ova could carry a mutation. If the F2 are examined, the number of mutagenized gametes is also two per F1. m, starting mutation; sup, suppressor mutation.
One consideration when choosing a starting mutation is to decide what type of mutation to suppress: a null mutation, a loss-of-function mutation, or a gain-of-function mutation. Suppression of a null mutation (X) will allow recovery of new mutations that allow bypass of the gene of interest. These will either be gain-of-function mutations of downstream genes in the same substrate dependent pathway (Y) or loss-of-function mutations of downstream negative regulators (W). However, suppression of a null mutation will not allow recovery of mutations in genes that directly interact with the starting gene's product (Figure 17).
On the other hand, suppression of a reduction-of-function mutation (X) will allow recovery of mutations in genes that interact with the gene of interest. A mutation in an interacting gene will be recovered if it removes the negative regulation of an upstream interacting gene (V). Mutations that allow bypass of the gene of interest will also be recovered (W and Y), as with using the null. Upstream genes that, when mutated, increase the activity of the loss-of-function mutation used will also be recovered (Z; see Figure 18).
Suppression of a gain-of-function mutation can allow the subsequent recovery of the opposite spectra of alleles as suppression of a loss-of-function mutation. For example, loss-of-function mutations of downstream genes will be recovered (Y) as well as gain-of-function mutations of downstream negativly regulated genes (W). These screens can be very powerful (for example, see Wu and Han, 1994 and Lackner et al., 1994) regarding suppression of the gain-of-function let-60 ras mutations; Riddle (1977) for suppression of dauer defective mutants; and Hodgkin (1986) for suppression of sex determination mutations). However, it is important to know that the starting mutation (X) increases normal gene activity and does not confer a novel activity (Figure 19).
For example, screens using an amber suppressible allele will not only yield suppressors of interest, but can also yield mutations in amber suppressor tRNAs. If the suppressors of interest occur at a very low frequency, the other more non-specific mutations may overwhelm the recovery of the desired mutations. Similarly, screens using alleles which are splice site mutations may yield mutations in splicing components as well as the suppressors of interest. Of course, it is not always possible to begin with knowledge of the exact defect of the mutation of interest; nonetheless one can test by construction whether a starting mutation is suppressed by known informational suppressors. e.g., sup-7 and smg-1 (see below for further details).
Analysis of suppressor mutations involves separating the suppressors of interest from intragenic revertants and informational suppressors, testing dominance, checking for phenotypes in an otherwise wild-type background, mapping the suppressor locus, and determining the spectrum of suppression. When a suppressor mutation is isolated, its only known phenotype is that it suppresses the original mutation. Thus, no assumptions can be made about the phenotype of the suppressor on its own; it could have an unexpected phenotype, be lethal, or be silent. Because of this, strains for the analysis of suppressor mutations must carry the starting mutation to allow scoring of the suppressor phenotype. In essence, the strain with the starting mutation is the background strain for further analysis. Besides this, analysis of suppressor mutations proceeds as for any other mutation. For example, to establish linkage with a recessive suppressor mutation, mut/mut; sup/sup animals can be crossed to mut/mut; marker/marker strains to construct a mut/mut; marker/+; sup/+ heterozygote, and linkage of sup and marker observed among the progeny (mut=starting mutation, sup=suppressor mutation, marker=any unrelated marker) (Figure 20A). Dominance can be tested by crossing mut/mut; sup/sup males to mut/mut; marker/marker hermaphrodites and examining the phenotype of the non-marker cross-progeny (Figure 20B). Similarly, complementation of recessive suppressor mutations can be tested by crossing sup-x/sup-x; mut/mut males to sup-y/sup-y; mut/mut; marker/marker hermaphrodites.
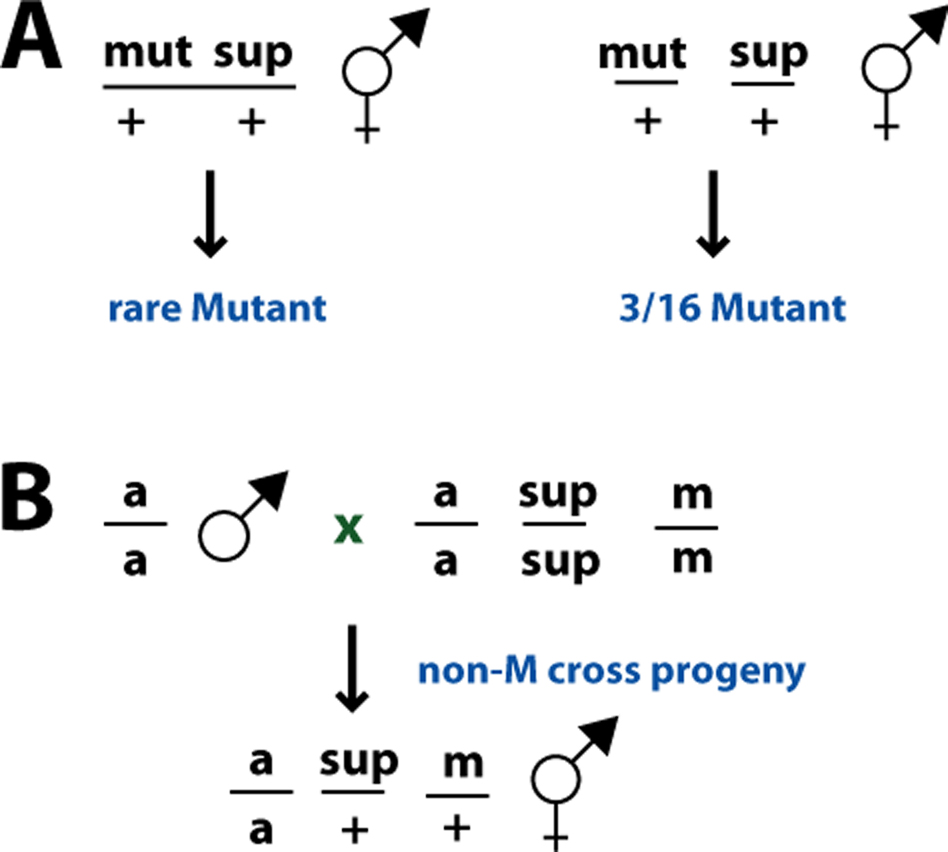 |
Figure 20. Analysis of suppressor mutations. (A) Linked versus unlinked suppressors. Assuming complete penetrance and recessivity of the sup mutation, only mut +/mut sup recombinants will be detected among the progeny of animals heterozygous for mut and sup. These will occur at a frequency of 2p/4 = p/2, where p is the map distance (see Sulston and Hodgkin (1988)). (B) Test for dominance. Animals heterozygous for the sup mutation and homozygous for the starting mutation, a, are constructed to determine the phenotype.
Intragenic revertants will be linked; extragenic linked suppressors can be separated by recombination. For analysis of intragenic revertants see Greenwald and Horvitz (1980). Such revertants can be informative with respect to the structure, function or expression of a particular gene, but are not helpful in defining a pathway.
It is important to determine the spectrum of suppression of a suppressor mutation: Does it suppress other alleles of the starting gene? If not, it might suggest informational suppression (see below) or a protein-protein interaction, depending on the nature of the alleles. Does it suppress hypomorphic or null alleles? If it only suppresses hypomorphic alleles, the suppressor might act upstream in a pathway or act in branch of the pathway. Does it suppress other phenotypes of the starting mutation? This would tell you if it acts in all pathways in which your starting mutation functions. Does it suppress mutations in other genes in the pathway, and if so, does it only suppress mutations that lie upstream? Not only can these types of data reveal whether a suppressor is informational, but they also indicate the specificity of the suppressor gene defined. Whether specific or generally acting suppressors are of greater interest is a matter of personal taste.
One class of extragenic suppressors are informational suppressors (reviewed by Hodgkin et al. (1987)). Formally, informational suppressors are allele-specific, gene non-specific, and thus act on particular types of mutations, rather than particular types of gene products. The classic informational suppressor is an amber suppressor, e.g., sup-7 in C. elegans. A new class of informational suppressors, the smg genes (e.g., smg-1), were found by reversion of three independent phenotypes; the smg genes affect mRNA stability (Pulak and Anderson, 1993). The informational nature of their suppression was discovered by the realization that three different genetic screens for unrelated pathways identified the same set of suppressor mutations (Hodgkin et al., 1989).
Once the map position of a suppressor mutation is determined, markers can be used in trans to separate the suppressor from the original mutation so that the phenotype of the suppressor alone can be determined. The simplest suppressor mutations to interpret are those that confer a phenotype opposite to the starting mutation, with suppression by virtue of epistasis [e.g., suppression of lin-15 multivulva by let-60 vulvaless (Han, et al., 1990); suppression of tra-3 masculinization by fem-1, fem-2 or fem-3 feminization (Hodgkin, 1986); suppression of dauer constitutive by dauer defective (Riddle, et al., 1981)]. However, many suppressor mutations have an unrelated phenotype or no phenotype other than suppression of the starting mutation; these are silent suppressors as they have no effect with respect to the phenotype of interest. There are various types of experiments one can carry out but no generalizable formula is available to determine what will be the most informative types of silent suppressor mutations. Note that silent suppressors can identify genes that would otherwise not have been identified.
Some suppressor mutations enhance other mutations that have the opposite phenotype to the starting mutation, suggesting that they indeed control activity of the pathway of interest. Some suppressor mutations may be due to a mutation in two genes and thus have a synthetic phenotype. For example, a double mutant animal carrying two suppressor mutations may display a phenotype opposite to the starting mutation and this suppression phenotype may not be seen unless both suppressor mutations are present. (In Figure 21 below, the mutation suppressed would be in the X gene; the redundant suppressors would be in the A and B genes.) The synthetic phenotype seen with the removal of both gene products (i.e., mutations in A and B in Figure 21) can be used for epistasis tests (i.e., with gene Y in Figure 21). If there is clear epistasis of the suppressor double mutant with mutations in the pathway, the redundant suppressors most likely act in a parallel subset of the otherwise linear pathway, with redundant genes A and B defined by suppressors of X (see Thomas et al. (1993) for an example). This pathway would look like: (Figure 21).
If suppressor mutations suppress hypomorphic alleles but not null alleles, then they are more likely to be a side branch of the main pathway. These mutations are in a side branch because they are unable to bypass the null allele, and thus do not define a downstream component regulated by an upstream gene (see Lee et al., 1994 for an example). Silent suppressors that do not show synthetic phenotypes cannot be placed in a pathway using epistasis, but the spectrum of suppression of mutations in the pathway of interest can suggest their role.
Transgenes add to the arsenal of tools for pathway analysis: they can be used for suppression and epistasis. For example, high copy number of lin-3 provided by a transgene results in a multivulva phenotype; this phenotype is hypostatic to the let-23 vulvaless phenotype, consistent with lin-3 acting upstream of let-23 (Hill and Sternberg, 1992). A powerful technique in the future will be reversion of the dominant effects of transgenes. One potential problem may be a high degree of intragenic revertants, that is, deletions of the integrated transgenes. In general this has not happened (Hajdu-Cronin et al., 1999, 2004; Hsieh et al., 1999).
In Saccharomyces cerevisiae, high copy suppression using plasmid libraries is a powerful tool for finding interacting or related genes. In C. elegans, similar analysis will be more difficult, but still possible. Due to the larger genome size compared to yeast, it would require testing on the order of 500 transgenic lines, each with multiple copies of five cosmids (assuming good representation of the genome with 2500 cosmids). The ability to create mutations in genes of interest will be a powerful tool. As genetically defined genes are cloned and homologies are seen, other molecularly defined components which may lie in the pathway of interest can be cloned through reverse genetics and engineered into a genetic mutation (Plasterk (1995); see Methods in cell biology for further details) for further analysis. Finally, from a geneticist's perspective, the genome project makes it more attractive to pursue the characterization of silent suppressor mutations because the molecular cloning will be increasingly facile.
The determination of the functional order of genes in a developmental pathway provides a working model for the design of additional experiments. New genes can be added to the pathway once they are found, either at a branchpoint or directly in the pathway. The gene order can be refined upon receipt of more information about the phenotype of the mutations involved, the isolation of new mutations that allow more epistasis analysis to be performed, as well as more information about the molecular and biochemical nature of the pathway of interest. Eventually, individual pathways will be connected to other pathways, evolving into a network of gene interactions for developmental processes.
The authors would like to thank Paul Garrity, Wendy Katz, Ralf Sommer, John DeModena, Marie-Anne Félix, Yvonne Hadju-Cronin, Howard Lipshitz, Katharine Liu, Anna Newman, and the editors for critical comments. Research in our laboratory discussed here was supported by the U. S .P. H. S, the March of Dimes Birth Defects Foundation, and the Howard Hughes Medical Institute, with which P. W. S. is an Investigator.
Ambros, V. (1989). A hierarchy of regulatory genes controls a larval to adult developmental switch in C. elegans. Cell 57, 49–57. Abstract Article
Ambros, V., and Moss, E.G. (1994). Heterochronic genes and the temporal control of C. elegans development. Trends Genet. 10, 123–127. Abstract Article
Anderson, P. (1995). Mutagenesis. Methods Cell Biol. 48, 31–58. Abstract
Aroian, R.V., and Sternberg, P.W. (1991). Multiple functions of let-23, a C. elegans receptor tyrosine kinase gene required for vulval induction. Genetics 128, 251–267. Abstract
Avery, L., and Wasserman, S. (1992). Ordering gene function: the interpretation of epistasis in regulatory hierarchies. Trends Genet. 8, 312–316. Abstract
Bargmann, C.I., and Avery, L. (1995). Laser killing of cells in Caenorhabditis elegans. Methods Cell Biol. 48, 225–250. Abstract
Chamberlin, H.M., and Sternberg, P.W. (1993). Multiple cell interactions are required for fate specification during male spicule development in Caenorhabditis elegans. Development 118, 297–323. Abstract
Clark, S.G., Lu, X., and Horvitz, H.R. (1994). The C. elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics 137, 987–997. Abstract
Edgley, M.L., Baillie, D.L., Riddle, D.L., and Rose, A.M. (1995). Genetic balancers. Methods Cell Biol. 48, 147–184. Abstract
Ellis, R.E., Jacobson, D.M., and Horvitz, H.R. (1991a). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics 129, 79–94. Abstract
Ellis, R.E., Yuan, J. and Horvitz, H.R. (1991b). Mechanisms and functions of Cell Death. Annu. Rev. Cell Biol. 7, 663–698. Abstract Article
Ferguson, E., and Horvitz, H.R. (1989). The multivulva phenotype of certain C. elegans mutants results from defects in two functionally-redundant pathways. Genetics 123, 109–121. Abstract
Ferguson, E.L., Sternberg, P.W., and Horvitz, H.R. (1987). A genetic pathway for the specification of the vulval cell lineages of Caenorhabditis elegans. Nature 326, 259–267. Abstract Article
Greenwald, I.S., and Horvitz, H.R. (1980). unc-93(e1500): a behavioral mutant of Caenorhabditis elegans that defines a gene with a wild-type null phenotype. Genetics 96, 147–164. Abstract
Hajdu-Cronin, Y.M., Chen, W.J., Patikoglou, G., Koelle, M.R., and Sternberg, P.W. (1999). Antagonism between G(o)alpha and G(q)alpha in Caenorhabditis elegans: the RGS protein EAT-16 is necessary for G(o)alpha signaling and regulates G(q)alpha activity. Genes Dev. 13, 1780–1793. Abstract
Hajdu-Cronin, Y.M., Chen, W.J., and Sternberg, P.W. (2004). The L-type cyclin CYL-1 and the heat-shock-factor HSF-1 are required for heat-shock-induced protein expression in Caenorhabditis elegans. Genetics 168, 1937–1949. Abstract Article
Han, M., Aroian, R., and Sternberg, P.W. (1990). The let-60 locus controls the switch between vulval and non-vulval cell types in C. elegans. Genetics 126, 899–913. Abstract
Hedgecock, E.M., Sulston, J.E., and Thomson, J.N. (1983). Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 220, 1277–1279. Abstract
Hengartner, M.O., Ellis, R.E., and Horvitz, H.R. (1992). Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 356, 494–499. Abstract Article
Hill, R.J., and Sternberg, P.W. (1992). The lin-3 gene encodes an inductive signal for vulval development in C. elegans. Nature 358, 470–476. Abstract Article
Hodgkin, J. (1986). Sex determination in the nematode Caenorhabditis elegans: analysis if tra-3 suppressors and characterization of fem genes. Genetics 114, 15–52. Abstract
Hodgkin, J., Kondo, K., and Waterston, R.H. (1987). Suppression in the nematode Caenorhabditis elegans. Trends Genet. 3, 325–329. Article
Hodgkin, J., Papp, A., Pulak, R., Ambros, V., and Anderson, P. (1989). A new kind of informational suppression in the nematode Caenorhabditis elegans. Genetics 123, 301–313. Abstract
Hsieh, J., Liu, J., Kostas, S.A., Chang, C., Sternberg, P.W., and Fire, A. (1999). The RING finger/B-box factor TAM-1 and a retinoblastoma-like protein LIN-35 modulate context-dependent gene silencing in Caenorhabditis elegans. Genes Dev. 13, 2958–2970. Abstract Article
Huang, L.S., Tzou, P., and Sternberg, P.W. (1994). The lin-15 locus encodes two negative regulators of C. elegans vulval development. Mol. Biol. Cell 5, 395–412. Abstract
Lackner, M.R., Kornfeld, K., Miller, L.M., Horvitz, H.R., and Kim, S.K. (1994). A MAP kinase homolog, mpk-1, is involved in ras-mediated induction of vulval cell fates in Caenorhabditis elegans. Genes Dev. 8, 160–173. Abstract
Lee, J., Jongeward, G.D., and Sternberg, P.W. (1994). unc-101, a gene required for many aspects of C. elegans development and behavior, encodes a clathrin-associated protein. Genes Dev. 8, 60–73. Abstract
Muller, H.J. (1932). Further studies on the nature and causes of gene mutations. Int. Congr. Genet. 6, 213–255.
Plasterk, R.H. (1995). Reverse genetics: from gene sequence to mutant worm. Methods Cell Biol. 48, 59–80. Abstract
Pulak, R., and Anderson, P. (1993). mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 7, 1885–1897. Abstract
Riddle, D.L. (1977). A genetic pathway for dauer larva formation in Caenorhabditis elegans. Stadler Genet. Symp. 9, 101–120.
Riddle, D.L., Swanson, M.M., and Albert, P.S. (1981). Interacting genes in nematode dauer larva formation. Nature 290, 668–671. Abstract Article
Sternberg, P.W. (1990). Genetic control of cell type and pattern formation in Caenorhabditis elegans. Adv. Genet. 27, 63–116. Abstract
Sternberg, P.W. (1990). Genetic control of cell type and pattern formation in C. elegans. Adv. Genet. 27, 63–115.
Sternberg, P.W. (1993). Intercellular signaling and signal transduction in C. elegans. Annu. Rev. Genet. 27, 497–521. Abstract Article
Sulston, J., and Hodgkin, J. (1988). Methods. In: "The Nematode Caenorhabditis elegans," W.B. Wood, ed. (Cold Spring Harbor Laboratory), pp. 587–606.
Tautz, D. (1992). Redundancies, development and the flow of information. Bioessays 14, 263–266. Abstract Article
Thomas, J.H., Birnby, D.A., and Vowels, J.J. (1993). Evidence for parallel processing of sensory information controlling dauer formation in Caenorhabditis elegans. Genetics 134, 1105–1117. Abstract
Tong, A.H., Lesage, G., Bader, G.D., Ding, H., Xu, H., Xin, X., Young, J., Berriz, G.F., Brost, R.L., Chang, M., et al. (2004). Global mapping of the yeast genetic interaction network. Science 303, 808–813. Abstract Article
Villeneuve, A.M., and Meyer, B.J. (1990). The regulatory hierarchy controlling sex determination and dosage compensation in Caenorhabditis elegans. Adv. Genet. 27, 117–188. Abstract
Wu, Y., and Han, M. (1994). Suppression of activated Let-60 Ras protein defines a role of C. elegans Sur-1 MAP kinase in vulval differentiation. Genes Dev. 8, 147–159. Abstract
*Edited by David Fay. WormMethods editor, Victor Ambros. Last revised April 22, 2005. Published May 16, 2006. This chapter should be cited as: Huang, L. S. and Sternberg, P. W. Genetic dissection of developmental pathways (May 16, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.88.1, http://www.wormbook.org.
§Adapted from: Huang, L.S., and Sternberg, P.W. (1995). Genetic Dissection of Developmental Pathways. In Methods Cell Biol. aka C. elegans: Modern Biological Analysis of an Organism, H.F. Epstein and D.C. Shakes, eds. (San Diego: Academic Press), 48, 97–122.
†To whom correspondence should be addressed. E-mail: linda.huang@umb.edu