Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergAbstract
Mutations in many genes can result in a similar phenotype. Finding a number of mutants with the same phenotype tells you little about how many genes you are dealing with, and how mutable those genes are until you can assign those mutations to genetic loci. The genetic assay for gene assignment is called the complementation test. The simplicity and robustness of this test makes it a fundamental genetic tool for gene assignment. However, there are occasional unexpected outcomes from this test that bear explanation. This chapter reviews the complementation test and its various outcomes, highlighting relatively rare but nonetheless interesting exceptions such as intragenic complementation and non-allelic non-complementation.
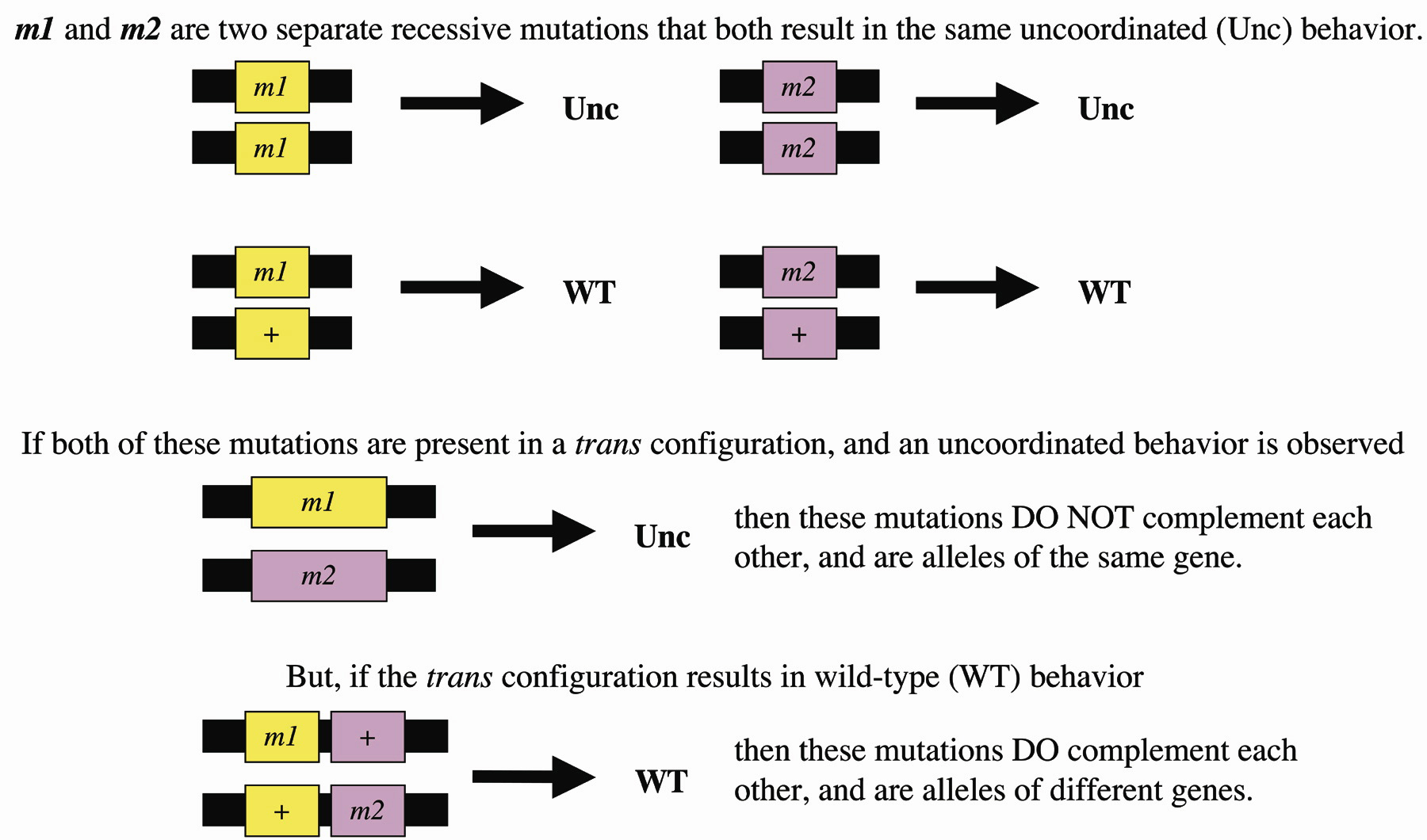
During the complementation test, if a phenotype is observed when a recessive mutation is combined in trans with another recessive mutation that has been mapped to the same area, it is concluded that these mutations are alleles of the same gene; neither allele produces a product that can restore wild-type function (Figure 1). However, if the double heterozygote exhibits a wild-type phenotype, it is concluded that the two mutations are alleles of different genes; each mutant has a functional version of the other gene. This assay requires that the mutation in question is genetically linked to the test mutation and that both mutations are recessive. The usefulness of this test can be most appreciated after large mutagenesis experiments, as demonstrated by Sydney Brenner in his pioneering mutagenesis screen in C. elegans. Through mapping and complementation tests, he was able to place 256 autosomal ethylmethane sulphonate (EMS) induced mutations into 77 complementation groups, making the genetic analysis of C. elegans much easier to approach (Brenner, 1974).
|
Figure 1. The complementation test. m1 and m2 are fictional alleles representing recessive mutations. Historical Note: The cis-trans test. In a more rigorous application of the complementation test; if the mutations do not complement one another when they are present in the trans configuration, the mutations should then be combined in cis and tested over the wild-type allele (Pontecorvo, 1958). In this case, the cis heterozygote should exhibit a wild-type phenotype. If a mutant phenotype is still observed, then the trans complementation test was leading you to the wrong conclusion. The correct conclusion is that the mutations do not define alleles of the same gene, rather they are exhibiting non-allelic non-complementation (see text). This result is rare and the cis test is not usually performed and may be quite difficult to carry out.
In essence, the complementation test is a very simple test and can quite satisfyingly give you an idea of the number of loci involved in a process after a successful mutagenesis experiment. Having said that, there are four considerations to keep in mind (Hawley and Walker, 2003). First, both mutations must be recessive. Second, the mutations can cause different homozygous phenotypes. Third, the trans-heterozygote can exhibit a more severe phenotype than either homozygote alone. Fourth, a mutation can affect more than one gene product in a complex locus.
Both mutations must be recessive. The complementation test will not work if either mutation exhibits dominance over the wild-type allele. In this case the phenotype of the dominant mutation will be exhibited whether or not the other allele is a mutation in the same gene; the mutations will appear to not complement. However, semi-dominant mutations can be put into complementation groups if the heterozygous phenotype is noticeably different from the homozygous phenotype (for examples see unc-58 and unc-93; Brenner, 1974; Greenwald and Horvitz, 1980; Park and Horvitz, 1986). Assigning a dominant mutation to a locus requires other methods (see Genetic suppression).
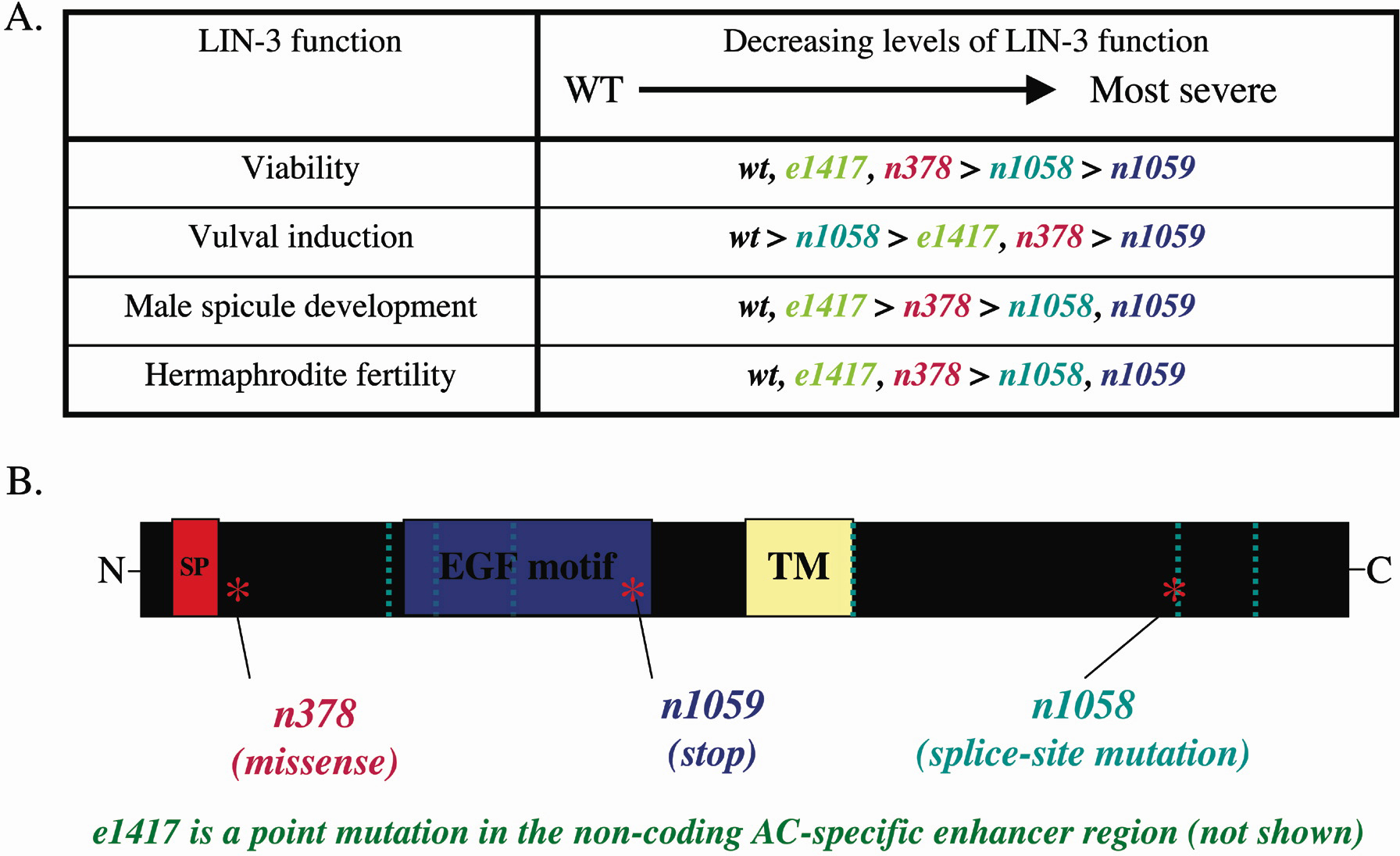
Mutations in the same gene can cause different homozygous phenotypes. If a gene functions in many processes, then mutations in this gene may impair each function independently. lin-3 is such a pleiotropic gene. lin-3 encodes the C. elegans member of the epidermal growth factor family and is an essential gene that is required in at least three different processes; vulval induction, male spicule development and hermaphrodite fertility (Ferguson and Horvitz, 1985; Hill and Sternberg, 1992; Liu et al., 1999). Mutations in lin-3 disrupt these functions to different degrees; homozygotes of one allele, e1417, are Vul whereas homozygotes of another allele, n1058, exhibit defects in male spicule development, hermaphrodite fertility, viability, and only very weakly, vulval induction (Figure 2A). Thus initially, it would appear that e1417 and n1058 are alleles of different genes. To further complicate the issue, these alleles exhibit complex complementation interactions (see Intragenic complementation below).
|
Figure 2. (A) LIN-3 is required in at least four processes, viability, vulval development, male spicule development and hermaphrodite fertility. lin-3 alleles exhibit a different allelic series for each process, suggesting that each process has a different, independently regulated requirement for LIN-3. Adapted from Liu et al., 1999. (B) Domain structure of LIN-3 and location of the lesions in lin-3 alleles. SP-signal peptide cleavage site, EGF-epidermal growth factor domain, TM-transmembrane domain, dotted vertical lines-intron boundries. Adapted from Liu et al., 1999. Information for e1417 is from Hwang and Sternberg, 2004.
The trans-heterozygote can exhibit a phenotype, that is more severe than either homozygote alone. This is also termed "negative complementation". This result contrasts with the expected phenotype of the trans-heterozygote having the phenotype of the weaker allele. Although rare, negative complementation has been noted for the sqt-1 locus. In this case, animals homozygous for sc100 or sc103 are wild-type, but sc101/sc103 male progeny have a weak Dumpy (Dpy) phenotype. Additionally, when sc100 is put in trans to a deficiency, the trans-heterozygotes have a weak Dpy phenotype (Kusch and Edgar, 1986). In general, mutations in many of the collagen genes result in interesting allelic interactions as discussed below.
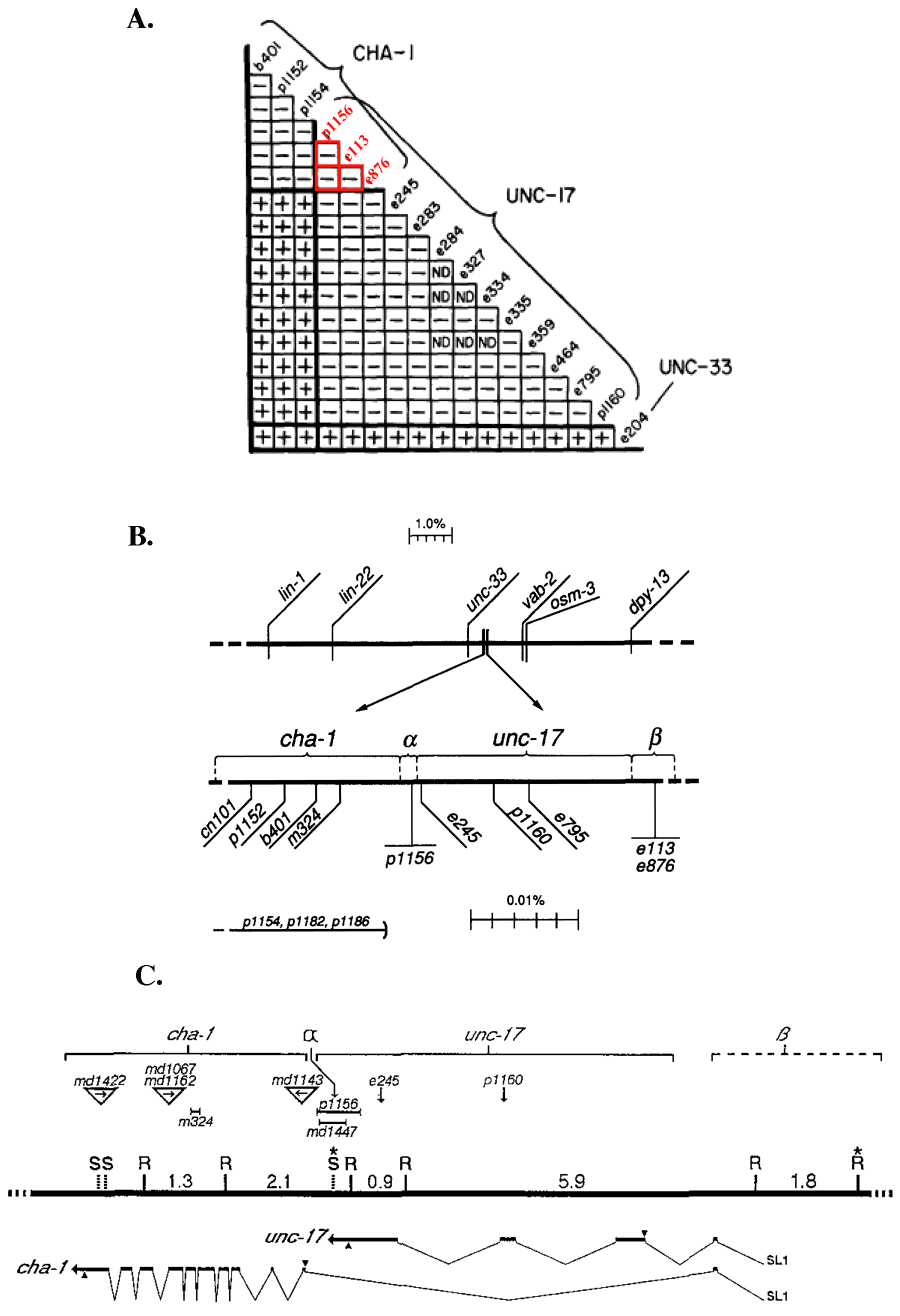
A mutation can affect more than one gene product if the genes are in a complex locus. A classic example of this type of genetic relationship is the cha-1/unc-17 locus (Figure 3). cha-1 and unc-17 are tightly linked and mutations in cha-1 and unc-17 result in very similar, severe behavioral and developmental defects (Rand and Russell, 1984; Rand, 1989). Although most alleles of unc-17 complemented cha-1, three hypomorphic unc-17 alleles failed to complement cha-1. Molecular characterization finally demonstrated that unc-17 and cha-1 encode independent mature transcripts produced by alternative splicing (Alfonso et al., 1993; Alfonso et al., 1994). The non-complementing mutations fall into two groups, α and β. The one α mutation is a deletion of the non-coding region between cha-1 and unc-17 that removes sequences required for unc-17 RNA processing and most likely for cha-1 expression and/or splicing. The two β mutations have yet to be identified, but they have been mapped to the shared 5' untranslated region of the locus and thus probably disrupt transcriptional regulation of both unc-17 and cha-1 (Alfonso et al., 1994). Thus the non-complementing alleles are actually alleles of both genes.
|
Figure 3. cha-1 and unc-17 form a complex locus. (A) The complementation matrix of alleles of cha-1 and unc-17. "+" indicates complementation, "—" indicates non-complementation. p1156, e113 and e876 do not complement alleles of cha-1 or unc-17 (boxed in red). unc-33(e204) was used as a complementation control. Reprinted with permission from Rand and Russell, 1984. Copyright ©1984 the Genetics Society of America. (B) p1156, e113 and e876 were fine mapped to two separate regions of the cha-1/unc-17 locus. p1156 defines the alpha region, to the left of the other unc-17 alleles, while e113 and e876 define the beta region, to the right of unc-17 alleles. Reprinted with permission from Rand, 1989. Copyright ©1989 the Genetics Society of America. (C) Alfonso et al. (1994) molecularly determined that unc-17 lies within the first intron of the cha-1 locus, the beta region is a non-coding region of the complex locus shared with cha-1, and the alpha region lies between the 3' end of unc-17 and splice site junction of cha-1. Top line corresponds to genetic boundaries between cha-1 and unc-17, with mutations noted. Middle line corresponds to genomic DNA with SalI (S) and EcoRI sites noted. Bottom line corresponds to cDNA structures of cha-1 and unc-17. Reprinted with permission from Alfonso et al. (1994), Copyright (1994), with permission from Elsevier. See reference for more details.
In summary, the complementation test is used to assign mutant alleles to specific genetic loci. Mutant alleles of the same gene fail to complement one another, while alleles of different genes do complement each other. In general, keeping in mind the considerations above, the interpretation of complementation test is quite straightforward. However, we are occasionally faced with complex patterns of complementation.
There are two ways the complementation test can mislead you. First, alleles of the same gene can sometimes complement each other, termed "intragenic complementation". Second, mutations of two separate genes can sometimes not complement one another, termed "non-allelic non-complementation". In both instances, complex allelic interactions are observed that can lead to problems knowing when and how to assign the mutant alleles to complementation groups. Although these allelic conundrums are found in many organisms, this discussion is focused on examples found in C. elegans.
During intragenic complementation, alleles of the same gene complement one another, even though both alleles produce a faulty gene product. There are different means by which mutant alleles of the same gene can mutually correct one another. First, one mutant gene product may reduce the dosage of the other mutant product. Second, a faulty complex formed by one mutant gene product may be stabilized by the presence of an alternatively mutant gene product. Third, a gene product containing a mutation that affects one function may provide the function missing from an alternatively altered gene product.
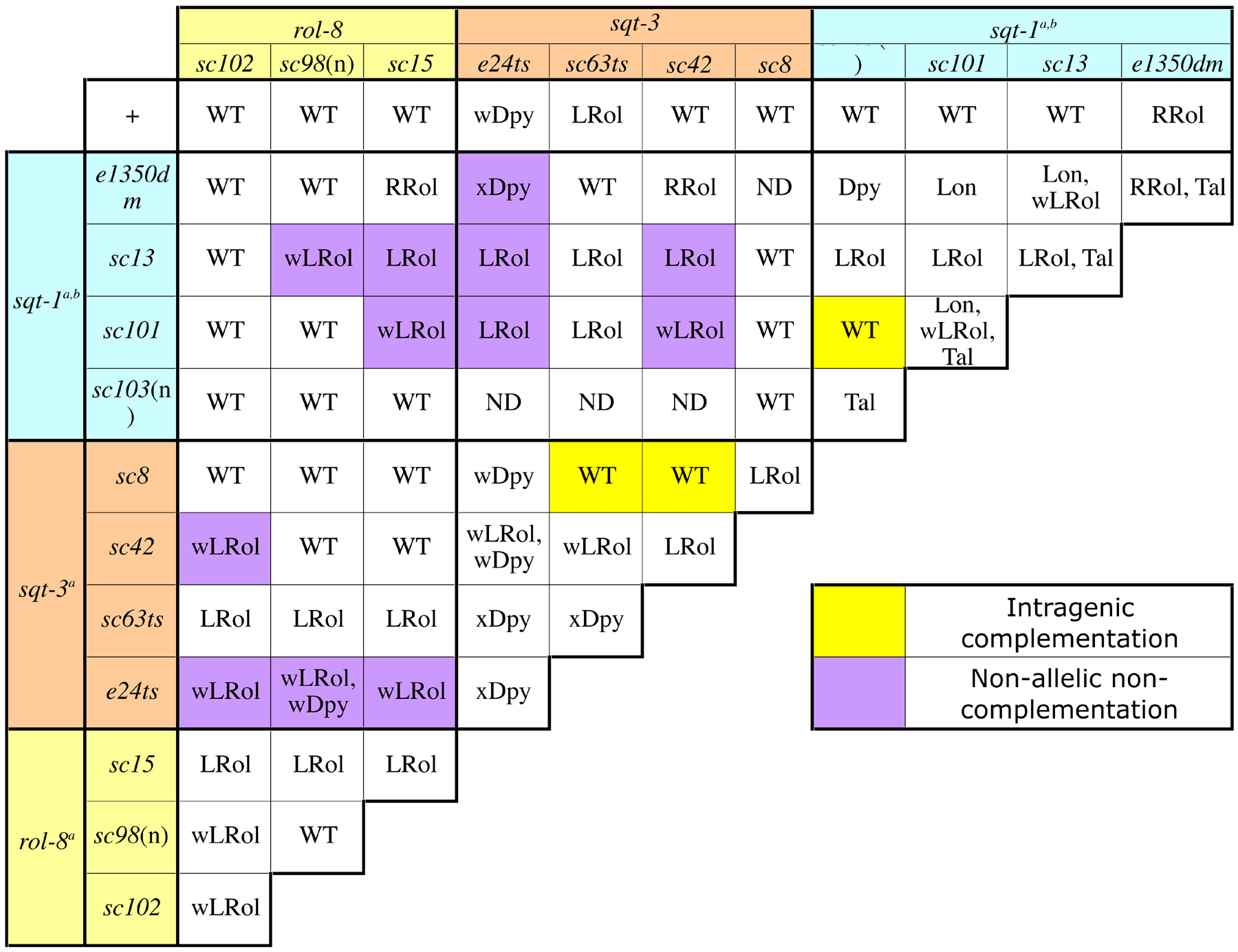
Alleles of genes required for cuticle formation exhibit intragenic complementation (De Melo et al., 2002; Kramer et al., 1988; Kusch and Edgar, 1986). In the case of the cuticle collagen sqt-1, the recessive missense mutation, sc101, is alleviated in trans by a null mutation, sc103 (Table 2 and Table 3; Kramer and Johnson, 1993). Although both sc101 and sc103 homozygotes have tail defects, and sc101/sc101 is a long weak left roller, the trans-heterozygotes are wild-type (Kusch and Edgar, 1986; Kramer and Johnson, 1993). This result suggests that the presence of the sc101 product has a negative effect on normal collagen associations, since decreasing the amount of the mutant product (e.g. replacing one allele with an allele that produces no product) restores a wild-type phenotype. The poisoning effect of the Gly-X-Y mutations seems to be especially detrimental to collagen proteins and cuticle formation as they are often found in alleles that exhibit non-allelic non-complementation with other collagen genes (see Non-allelic non-complementation).
Table 1. Phenotypes of lin-3 homozygous and heteroallelic strains
| lin-3 allele | n378 | e1417 | n1058 | n1059 |
| n378 | 97% Vul (n=266) | 88% Vul (n=226) | 78% Vul (n=364) | 100% Vul (n=665) |
| e1417 | 89% Vul (n=351) | 59% Vul (n=414) | 99.8% Vul (n=471) | |
| n1058 | Sterile; occasional arrested larvae | Arrested larvae | ||
| n1059 | Arrested larvae | |||
| lin-3 alleles exhibit intragenic complementation. e1417/n1058 and n378/n1058 trans-heterozygotes are viable and are less likely to be Vul than e1417 or n378 homozygotes. Abbreviations:Vul-Vulvaless. Reprinted with permission from Ferguson and Horvitz, 1985. Copyright ©1985 the Genetics Society of America. | ||||
Table 2. Mutated domains of the non-allelic, non-complementing SQT gene products
| Allele | Collagen domain distrupted | References | |
|---|---|---|---|
|
sqt-1 |
e1350dm | N-terminal propeptide cleavage site | |
| sc13 | Cysteine domain /C-terminal propeptide cleavage site | ||
| sc101 | Gly-X-Y repeat | ||
| sc103(n) | All (N-terminal stop) | ||
|
sqt-3 |
sc8 | C-terminal propeptide cleavage site | |
| sc42 | Signal peptide | ||
| sc63ts | Gly-X-Y repeat | ||
| e24ts,sd | Gly-X-Y repeat | ||
|
rol-8 |
mutations not determined |
Table 3. Complex interactions are observed between collagen genes, sqt-1, 2, 3 and rol-8
|
| For sqt-3 interactions, trans-heterozygotes were assayed at 20° and 25° C. Only the results from the 25° assay are included here. See references for more instances of non-allelic non-complementation. In some instances, complementation was also observed among alleles of the same gene (highlighted in yellow). Abbreviations are as follows. (n)-null allele, RRol-Right Roller, Lon-Long, Dpy-Dumpy, LRol-Left Roller, Rol-Roller, Tal-abnormal tail, ND-not discussed. Modifiers; x-extreme, w-weak. a Data from Kusch and Edgar (1986). b Data from Kramer and Johnson (1993). |
In 1964, Crick and Orgel proposed that inter-allelic complementation may be due to a "good corrects bad" mechanism (Crick and Orgel, 1964). In this scenario, a mutation in a subunit of a homomer or multimer would result in the mis-folding of the monomer subunit and disrupt the activity of the whole complex. Likewise, a subunit with a different mutation would also produce a faulty complex. However, a functional complex can be restored if one mutant subunit is able to structurally compensate for the other mutant subunit.
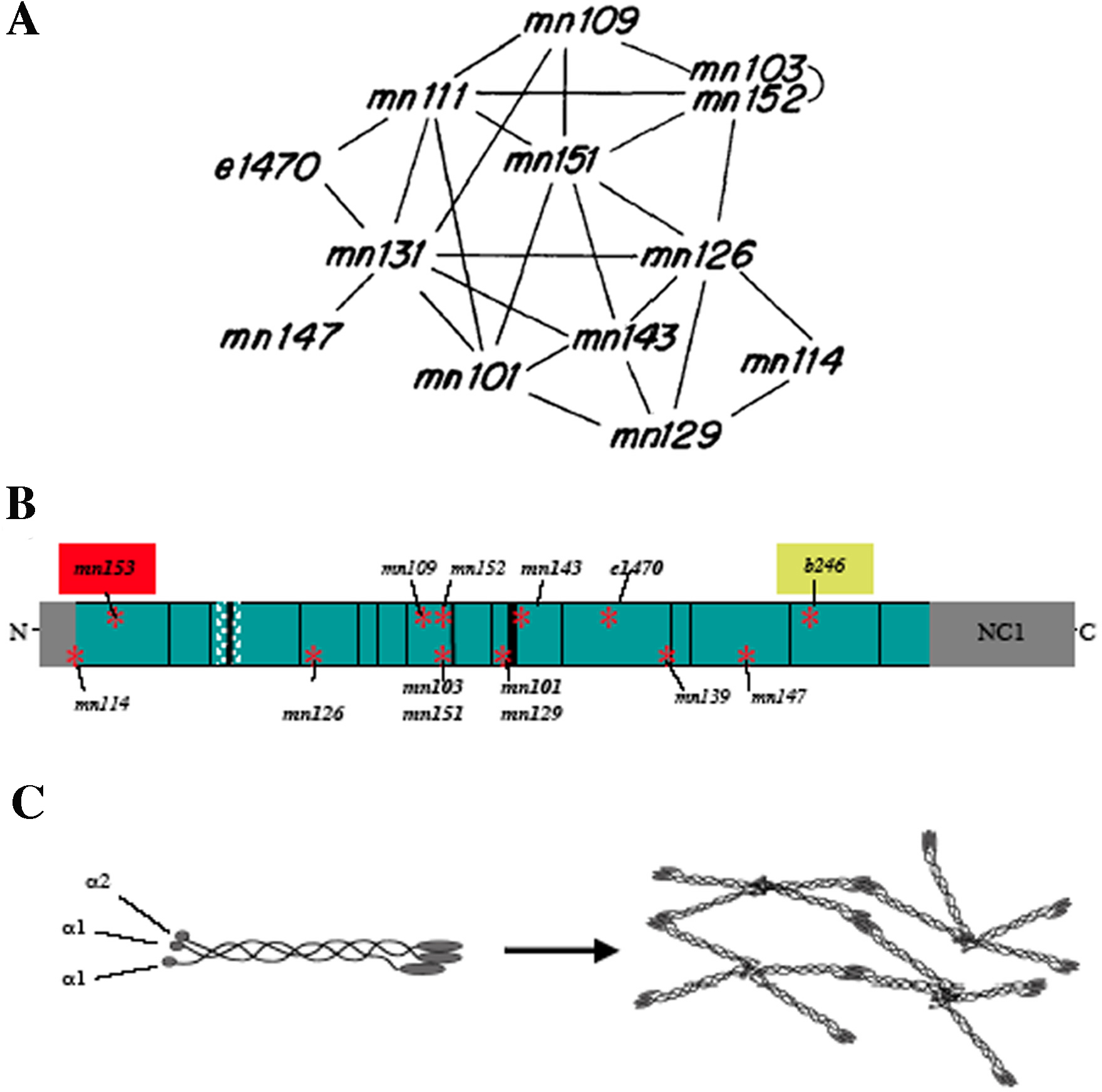
One example of this type of mutual correction may be demonstrated by alleles of let-2. The initial identification of let-2 occurred in a screen for X-linked lethals and steriles (Meneely and Herman, 1979). In this screen, four alleles of let-2 were identified and were shown to exhibit a complex complementation pattern. In fact, every allele complemented at least one other allele, including two alleles identified independently in other labs. A second screen published a few years later identified another eight alleles, which also exhibited the same complex pattern, giving rise to a complicated web of allelic interactions (Figure 4A; Meneely and Herman, 1981). These alleles also complement at least one other allele (see Table 4; Meneeley and Herman, 1981). The strongest allele, mn153, is a mutation in an N-terminal splice site and does not complement any other allele except b246. b246 exhibits the least severe phenotype of all the alleles; it is embryonic lethal only at non-permissive temperatures. Unexpectedly, b246 complements all other let-2 alleles (see Table 4). let-2 encodes an α2(IV) collagen chain, which is a major component of basement membrane (see Basement membranes; Sibley et al., 1993). Like other collagen molecules, LET-2 has an extensive Gly-X-Y repeat domains, making up about three-quarters of the molecule, and conserved non-collagenous regions (Figure 4B). Type IV collagen molecules are made up of heterotrimers of one α2(IV) chain and 2 α1(IV) chains (Figure 4C). These heterotrimers dimerize via their conserved C-terminal NC1 domains and can tetramerize via their C-termini to form a collagen lattice that provides structural support for basement membranes. The strong mutations in the Gly-X-Y regions can disrupt trimer formation and cause the accumulation of misfolded heterotrimers in the cell in a temperature dependent manner (Gupta et al., 1997). The b246 mutation occurs in a region of relatively high thermal stability and is therefore unlikely to have as drastic an effect on triple helical formation as mutations in regions of lower thermal stability (such as the mutations noted in bold in Figure 4B) in subsequent complex formations (Sibley et al., 1993). The presence of the b246 mutant product in the type IV collagen heterotrimers may help to stabilize the presence of other more severely altered heterotrimers in trans-heterozygotes.
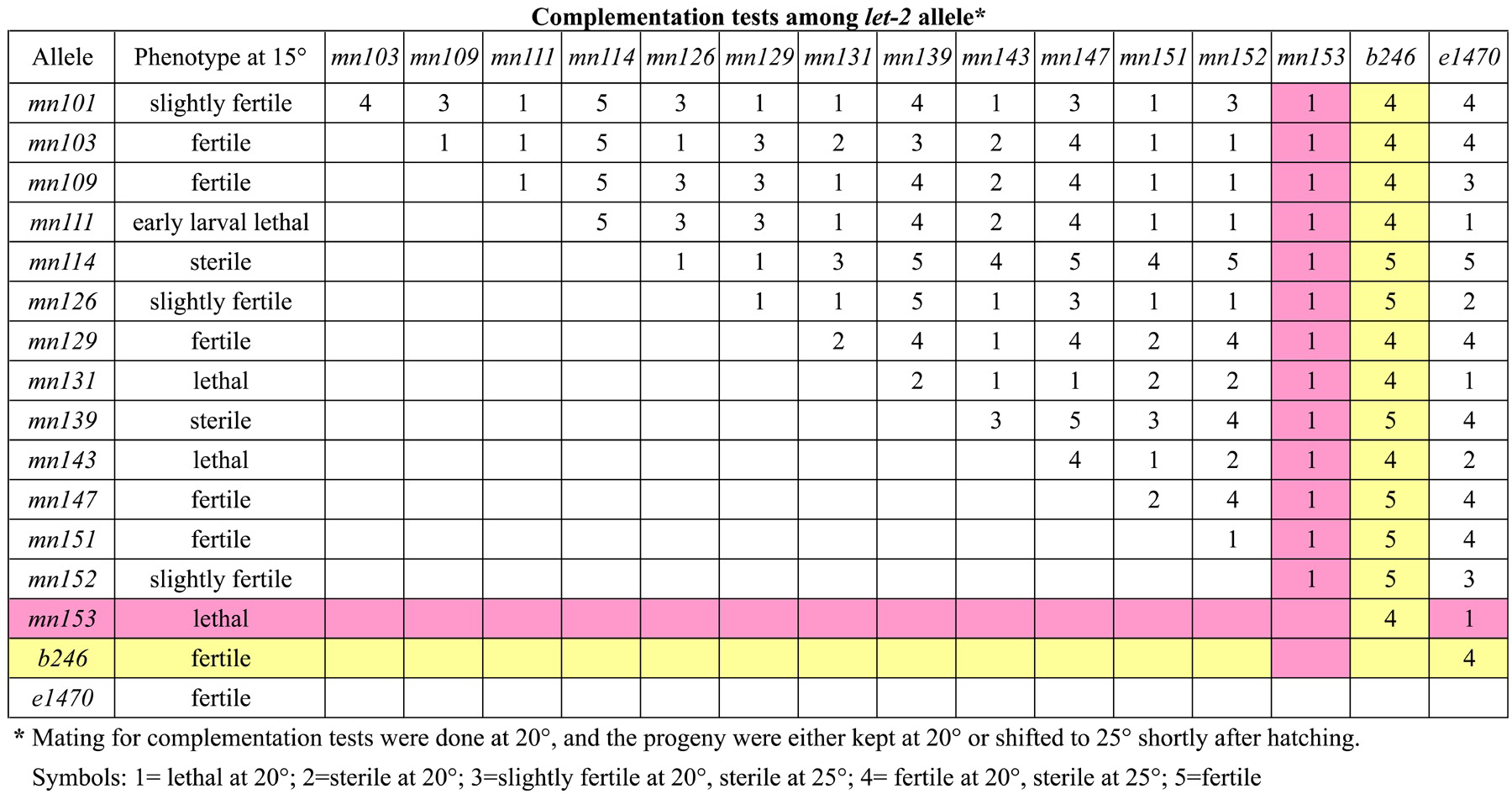
Table 4. Complementation tests among let-2 allele*
|
| Table of complementation interactions among the let-2 alleles, including mn153, which does not complement any of the other alleles except b246, which complements every allele. Reprinted with permssion from Meneely and Herman, 1981. Copyright ©1981 the Genetics Society of America. |
|
Figure 4. Intragenic complementation of let-2 alleles. (A) let-2 alleles exhibit a very complex pattern of complementation. Lines between alleles indicate non-complementation (the expected outcome). Alleles that are not joined by a line complement one another to some extent. Reprinted with permission from Meneely and Herman, 1981. Copyright ©1981 the Genetics Society of America. (B) A schematic of the LET-2 (α2)IV collagen, adapted from Sibley et al. (1993). Conserved type IV collagen features include extensive Gly-X-Y repeats (in blue) with interspersed interruptions (vertical black lines), non-collagenous termini (in grey) with a conserved NC1 domain at the C-terminus. Cross-hatched area denotes an alternative splice region. Locations of mutations are indicated with the strongest mutations (>90% homozygous embryonic lethality at 20°) in bold. mn153 (boxed in red), is the only mutation that does not complement any other allele aside from b246 (boxed in green), which complements all other alleles to some extent. The interruptions in the Gly-X-Y repeat domains lower the thermal stability of the region resulting in more severe phenotypes of Gly-X-Y mutations (noted in bold). (C) Type IV collagen is composed of a heterotrimer of 1 α2(IV) collagen chain and 2 α1(IV) chains. These heterotrimers can dimerize at their C-terminal NC1 domains, and then form a complex lattice through tetramerization of their N-terminal regions and lateral interactions along their triple helical domains. Mutations in the Gly-X-Y repeats would disrupt heterotrimer formation and subsequent associations.
Many examples of intragenic complementation have been reported for genes that encode products with independently functioning domains, or loci that are involved in independently regulated functions. In these cases, a mutation can result in a gene product that specifically disrupts one process while functioning relatively normally in others. Trans-heterozygotes made with such alleles exhibit intragenic complementation. This appears to be the most frequently reported type of intragenic complementation in C. elegans, perhaps since it results in some very interesting interactions. This type of intragenic complementation has been observed for a number of genes, including lin-3, gld-1, unc-5, unc-84, and bli-4.
As mentioned before, lin-3 is required in different developmental processes. lin-3 is also independently mutable, such that a mutation in one part of the gene can disrupt one process while having minimal effects on other LIN-3 requiring processes. For example, the e1417 mutation only disrupts vulval induction, having no effect on male spicule development, while the n1058 mutation only weakly disrupts vulval induction, but strongly disrupts male spicule development (Liu et al., 1999). These homozygous phenotypes suggest that each allele has residual function in certain tissues. Furthermore, the n1058 mutation can complement two other lin-3 alleles, e1417 and n378, for vulval induction (see Table 1 and Table 3 in Liu et al., 1999). The intragenic complementation in this case suggests that residual function from each allele is enough to complement the missing tissue specific function of the other allele, especially since the null mutation, n1059, does not complement any of these alleles (Table 1). Molecular analysis of the lin-3 mutations supports these genetic observations. Specifically, e1417 has a mutation in an anchor-cell specific enhancer, which is required for LIN-3 function in vulval development, but would not affect LIN-3 function in other tissues (Hwang and Sternberg, 2004; Figure 2B). Additionally, e378 has a missense mutation resulting in reduction-of-function and n1058 has a splice site mutation, which may cause a truncated LIN-3 product or lowered amount of the LIN-3 product to be made (Ferguson and Horvitz, 1985; Hwang and Sternberg, 2004; Liu et al., 1999). Neither of these mutations would completely remove LIN-3 function.
Such functional compensation has also been observed for alleles of gld-1, unc-5 and unc-84 (Francis et al., 1995; Merz et al., 2001; Malone et al., 1999). For example, gld-1 encodes an RNA binding protein required for several aspects of cell cycle progression during gametogenesis (Jones and Schedl, 1995). GLD-1 is required in a temporally and spatially regulated manner during germline development, and these requirements are reflected in the range of mutant phenotypes exhibited by gld-1 mutants. gld-1 mutations fall into five classes, A-E (Francis et al., 1995). Class C mutations result in the masculinization of the germline so that only sperm and no oocytes are produced. gld-1 class D mutations result in the opposite phenotype of feminization of the germline so that only oocytes and no sperm are produced. Trans-heterozygotes between class C and D alleles produce a wild-type phenotype presumably because gene products carrying the class C mutation can provide GLD-1 function during spermatogenesis while products carrying the class D mutation provide GLD-1 function during oogenesis.
Finally, if a mutation results in the lack of expression of gene product in a subset of cells, a compensating mutation can be one that restores that expression pattern. One example of compensation by expression is illustrated by mutations of bli-4. bli-4 encodes an essential Kex2/substilin-like protease that cleaves the N-terminus of pre-pro-peptides (Thacker et al., 1995). Two lethal alleles of bli-4, sy90 and h754, which do not complement other lethal alleles of bli-4, were found to complement the viable Blister allele, e937 (Peters et al., 1991; Rose and Baillie, 1980). Other lethal alleles exacerbate the Blister phenotype of e937. bli-4 encodes nine splice variants of the Kex2/substilin-like protease. All variants share the first 12 exons, including the coding region for the protease domain. Mutations in the protease domain affect all nine isoforms and result in embryonic arrest. e937 is a deletion that affects 5 of the 9 isoforms (Thacker et al., 2000). The sy90 and h754 mutations have not been identified, suggesting that these mutations may lie in regulatory elements. In addition, sy90 and h754 homozygous mutants arrest development during L1, which is later than the other lethal mutants suggesting that these mutations alter expression or function of BLI-4 after the embryonic period. Thus in the e937/sy90 or e937/h754 trans-heterozygote it appears that enough BLI-4 is supplied by the e937 allele to proceed past L1 development while the sy90 or h754 alleles may provide enough BLI-4 in later stages to abrogate the Blister phenotype.
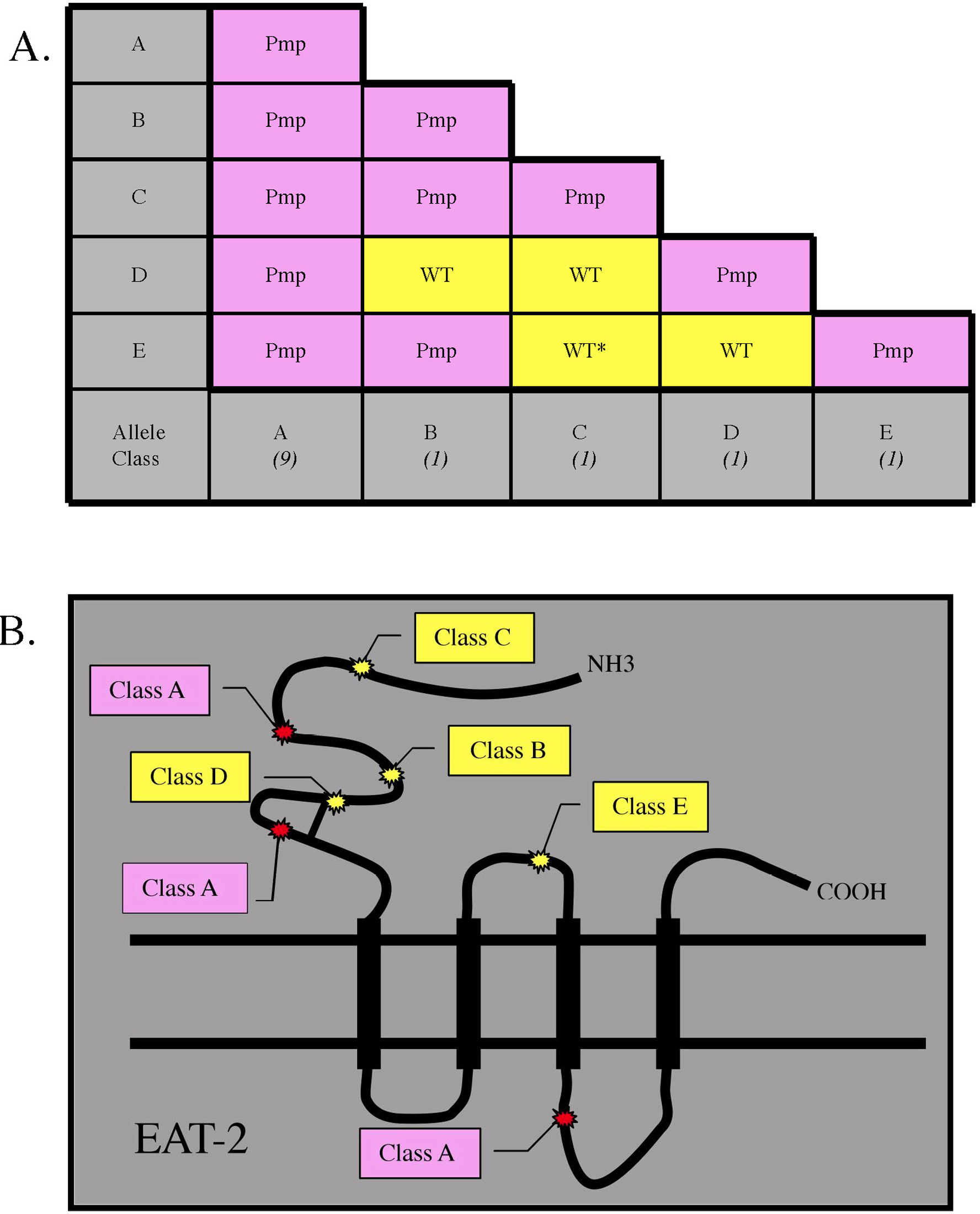
One last example of intragenic complementation, which could occur through stabilizing a complex, or by providing a compensatory function, is exhibited by eat-2 alleles. eat-2 encodes a subunit of the nicotinic acetylcholine gated ion channel required for pharyngeal pumping (Raizen et al., 1995). Nicotinic acetylcholine gated ion channels are hetero-pentamers that are activated by endogenous acetylcholine or drugs such as nicotine. The extracellular ligand binding sites are formed at the interfaces between two different subunits (Changeux and Edelstein, 1998). Alleles of eat-2 fall into five classes, A-E. Although class A alleles do not complement alleles in any class, alleles in class B-E exhibit intragenic complementation (Figure 5A). The eat-2 mutations were sequenced and complementing mutations of class B-E were located in the extracellular domain of the subunit (Figure 5B; McKay et al., 2004). In general, disruptions in extracellular domains would be expected to alter ligand-binding sites. One can imagine then that a channel composed of either faulty subunit would not form any stable ligand binding sites; however, two differently altered subunits might stabilize the channel and the ligand binding sites to allow some increase in channel activity. Alternatively, in an eat-2 trans-heterozygote with two faulty EAT-2 subunits, two different channels might be made with compensatory ligand specificities, thus restoring the functions of the EAT-2 nicotinic acetylcholine receptor.
|
Figure 5. (A) Intragenic complementation of eat-2 alleles. The number of alleles in each class is noted in parentheses below the class letter along the bottom row. Intragenic complementation results are highlighted in yellow. *These trans-heterozygotes are not fully wild-type; however, thay are less defective than either homozygote. Data was extrapolated from Raizen et al. (1995). (B) A schematic of the EAT-2 subunit with approximate locations of the mutations that can complement other eat-2 mutations (highlighted in yellow). Class A non-complementing mutations are highlighted in purple. Data extrapolated from McKay et al. (2004). Abbreviations: Pmp-Pumping defective.
Non-allelic non-complementation, also referred to as second-site non-complementation†, intergenic non-complementation, unlinked non-complementation, or second-site dominant enhancer, occurs when alleles of two different loci behave as if they are alleles of the same locus, e.g. the double heterozygote m1/+; +/m2 looks like either homozygote m1 +/m1 + or + m2/+ m2. Such interactions are not routinely encountered during complementation testing, because such assays are usually done between mutations that have been mapped to the same region and thus have a physical connection on the chromosome. By contrast, this interaction between mutations does not result from their physical location in the genome, but instead reflects a functional connection between different gene products, and often implies that the gene products physically interact. Other experimental approaches may however reveal that this effect is not uncommon. Specifically, screens for new alleles of a gene have the potential to uncover non-allelic non-complementing mutations (Hays et al., 1989). Hawley and Walker extensively review second-site non-complementation giving in depth examples of this event found in Drosophila and other organisms (Hawley and Walker, 2003). The following examples from C. elegans complement their review. Non-allelic non-complementation can be due to two situations: First, one or both mutations act as poisons to protein complexes, thus functional protein complexes become a limiting factor in a process. Second, both mutations reduce a threshold level of gene product, thus the dosage of the individual gene product becomes a limiting factor in a process. These situations, referred to as The Poison and Dosage Models, were put forth by M. Fuller, T. Stearns and D. Botstein and are discussed in more detail below (Fuller et al., 1989; Stearns and Botstein, 1988).
In the Poison Model of non-allelic non-complementation, an altered gene product impairs the protein complex with which it normally associates. Although the complex-poisoning effect of this mutation does not result in a visible phenotype on its own in heterozygotes, a simultaneous mutation in another member of the protein complex can reveal a visible defect. Such interactions have been observed between α and β tubulin genes in Drosophila and yeast, where altered a tubulin act as poisons by either sequestering β tubulin or by disrupting the polymerization of the microtubule (Fuller et al., 1989; Hays et al., 1989; Stearns and Botstein, 1988).
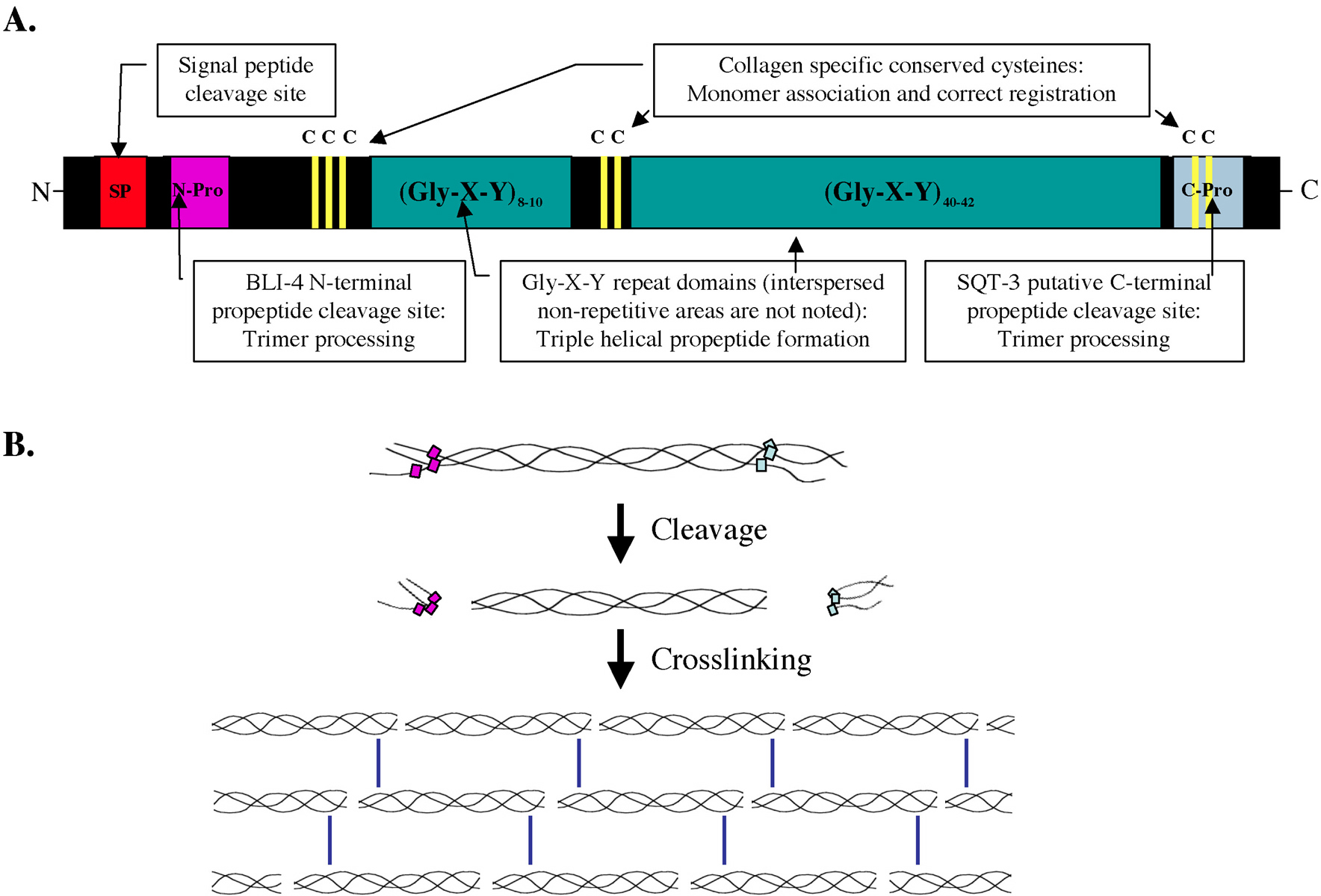
Such poisonous interactions have also been observed in C. elegans; most notably, Kusch and Edgar observed many instances of unconventional allelic interactions in their study of mutations affecting body shape and morphology (Kusch and Edgar, 1986). Specifically, it was noted that sqt-1 alleles did not complement certain sqt-3 or rol-8 mutations (Table 3). sqt-1, sqt-3 and rol-8 all encode cuticle collagens (Cox et al., 1989; Kramer et al., 1988; Novelli et al., 2004; van der Keyl et al., 1994). Collagens undergo significant amounts of homo- and heteromeric interactions, propeptide processing and crosslinking that result in permanent associations (Figure 6B; Johnstone, 2000; Myllyharju and Kivirikko, 2004; Page and Winter, 2003). Thus, an aberrant collagen monomer can potentially disrupt the cuticle matrix at many stages of cuticle assembly. The prevalence of non-allelic non-complementation among alleles of cuticle collagen genes supports this notion. In addition, mutations among collagen alleles that exhibit non-allelic, non-complementation interactions tend to disrupt a Gly-X-Y repeat domain, which is proposed to be required for monomer associations (Table 2, Figure 6; Kramer and Johnson, 1993; Novelli et al., 2004; van der Keyl et al., 1994). This suggests that mutations in the Gly-X-Y repeats act as poisons by causing disruptions in trimer formation and by sequestering wild-type monomers in non-functional protein interactions thereby decreasing the amount of functional trimers.
|
Figure 6. Cuticle collagens have a conserved domain structure. (A) Collagens in vertebrates have been demonstrated to form propeptide homo- or heteromeric helical trimers. Trimer formation is preceded by the association of the cysteine containing domains that are specific for each family of collagen. The association of the cysteine domains also puts the monomers in the right registration for trimer formation, which is carried out by the Gly-X-Y repeats in a zipping fashion starting from the C-terminal end. (B) The helical trimer propeptide is further processed by protease cleavage of the N-terminus by BLI-4 (in SQT-3 the C-terminus may also be a site of propeptide cleavage; Novelli et al., 2004). After proteolytic processing, the trimers are cross-linked to other trimers via N-terminal and C-terminal cross-linking sites to form the collagen matrix.
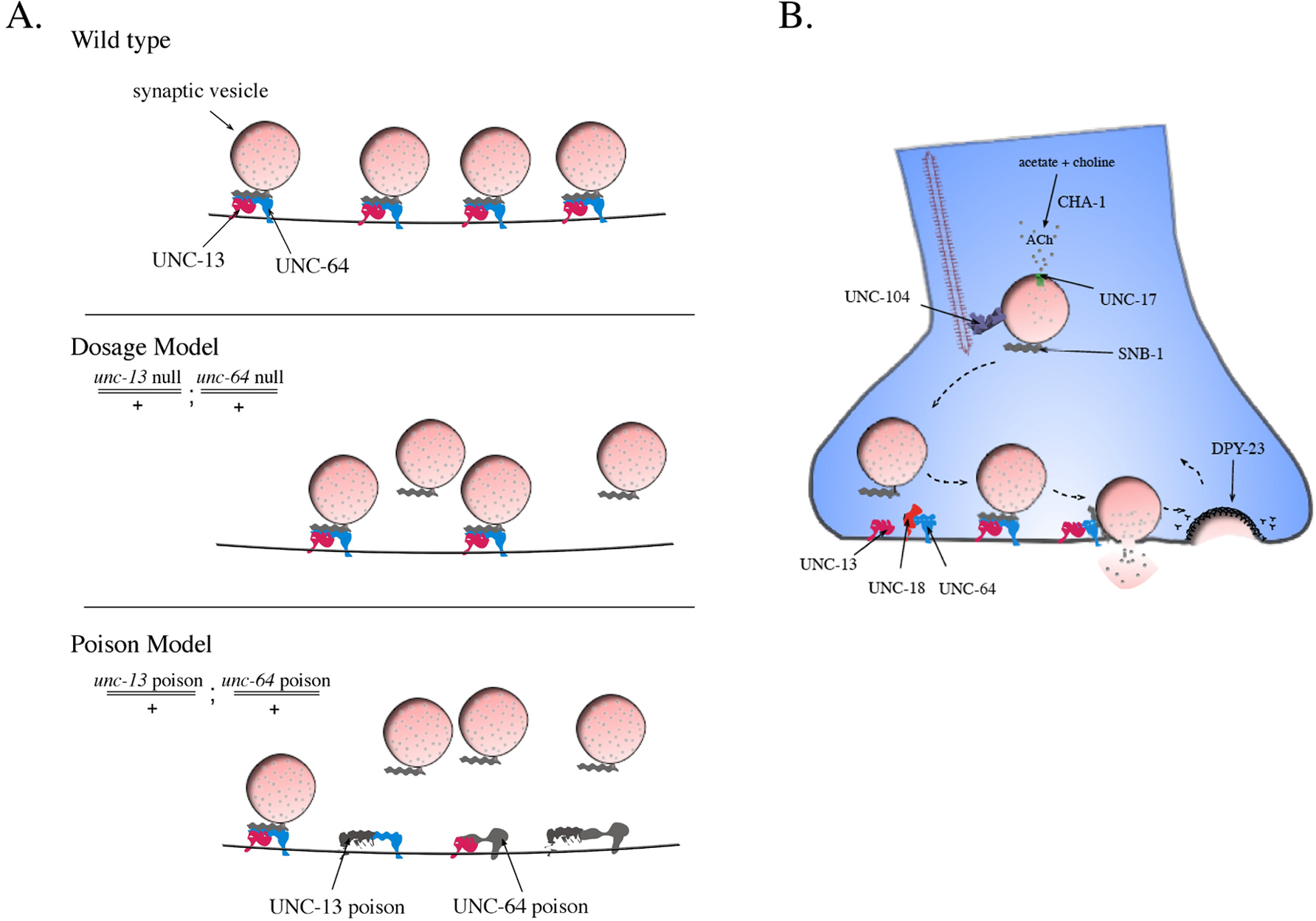
Non-allelic non-complementation has also been observed among genes required for synaptic vesicle fusion (Yook et al., 2001). In particular, a hypomorphic allele of unc-13, n2813, acts as a poison in synaptic transmission. unc-13 (n2813) is a recessive allele that causes a jerky, Unc phenotype due to decreased synaptic transmission. However, n2813 as a trans-heterozygote with mutations in other synaptic function loci exhibits an Unc phenotype even when wild-type copies of both loci are present (E. Jorgensen, pers. comm.). Furthermore, in a sensitive drug assay, n2813 exhibits a poisonous effect on synaptic transmission. UNC-13 is a diacylglycerol binding protein with multiple C2 Ca++ binding domains that physically interact with UNC-64/syntaxin to prime vesicles for fusion to the plasma membrane (Ahmed et al., 1992; Maruyama and Brenner, 1991; Richmond and Broadie, 2002). It was demonstrated that unc-13(n2813) did not complement a null or hypomorphic allele of unc-64, although a null allele of unc-13 fully complemented the unc-64 null allele. These results suggest that synaptic transmission is not sensitive to the amount of its individual UNC-13 or UNC-64 protein molecules; however, the number of functional UNC-13/UNC-64 protein complexes is a limiting factor. It is possible that UNC-13 (n2813) acts as a poison to synaptic transmission by sequestering wild-type UNC-64 into non-functional complexes, thus making the amount of UNC-64 a limiting factor to synaptic transmission, following the Poison Model of non-allelic non-complementation (Figure 7A).
|
Figure 7. The Poison and Dosage Models of vesicle priming at the synapse. (A) A schematic of primed vesicles along the active zone at a wild-type synapse. In The Dosage Model, the unc-13(null)/+ ; unc-64(null)/+ trans-heterozygote would have half the amount of UNC-13 and UNC-64, which would result in a decrease in the number of primed vesicles ready for synaptic release. In The Poison Model, the unc-13(poison)/+; unc-64(poison)/+ trans-heterozygote would produce products that would interfere with the formation of functional complexes. Altered UNC-13 and altered UNC-64 can participate in the same complex and render it non-functional. In addition these altered products can form complexes with wild-type partners, sequestering them in non-functional complexes. These interactions would reduce the level of functional complexes to below half the normal amount. (B) The synaptic vesicle cycle at a cholinergic synapse. Synaptic vesicles and associated proteins are transported to the synapse from the cell body by the synaptic vesicle kinesin protein UNC-104. Acetylcholine, made by CHA-1, is packaged into vesicles by the acetylcholine transporter, UNC-17. Mature synaptic vesicles must be docked and primed at the synapse so that the vesicle can rapidly fuse with the plasma membrane when the neuron is depolarized. Docking and priming of the vesicle requires the dissociation of UNC-18 from UNC-64/syntaxin and the association of UNC-13 with UNC-64. The vesicle is fully primed when SNB-1 joins the UNC-13/UNC-64 complex. After vesicle fusion, the vesicle and its associated proteins are recovered from the plasma membrane through clathrin-mediated endocytosis, which utilizes the μ2/DPY-23 containing AP-2 adaptor complex (W.S. Davis et al., WBPaper00023077). Recycling of the synaptic vesicles and their associated proteins is required for maintaining a readily releasable pool.
These results support the view that non-allelic non-complementation signifies a physical interaction between the mutant gene products; however, in the same study it was demonstrated that this is not always the case. Specifically, unc-13 (n2813) failed to complement mutations in other genes required for synaptic vesicle dynamics (Figure 7B). Whereas the greatest degree of non-complementation occurred between mutations in genes whose products are known to have a physical interaction (i.e. unc-13 and unc-64, unc-13 and unc-18, unc-64 and snb-1), non-complementation was also observed between genes whose products are not known to have a physical interaction (i.e. unc-13 and snb-1, unc-13 and dpy-23, unc-13 and unc-104). These observations suggest that the unc-13(n2813) aberrant gene product makes synaptic transmission sensitive to perturbations in other synaptic function loci. In particular, the effects of the UNC-13 poison extends to those loci that affect the concentration of synaptic components at the synapse, such as unc-104 and dpy-23. However, there is a limit to this interaction as non-complementation was not observed for unc-13 and cha-1, unc-13 and unc-17 or unc-13 and syd-1 trans-heterozygotes, as cha-1, unc-17 and syd-1 do not play a direct role in the synaptic vesicle cycle (see references in Yook et al., 2001).
In the Dosage Model, the limiting factor in a process is the total amount of gene product such that a simultaneous decrease in the levels of expression of both genes results in a mutant phenotype. Dosage-sensitive processes have been reported for developmental pathways where events are controlled by protein gradients, such as observed in Drosophila and vertebrates (Jackson and Berg, 1999; Kidd et al., 1999; Rancourt et al., 1995). Few examples of dosage sensitive processes have been reported in C. elegans; however, combined-haplo-insufficiency has been reported among ram genes required in male tail ray morphogenesis (Baird and Emmons, 1990). Baird and Emmons demonstrated that null-like mutations in ram-4 do not complement presumed null mutations in any of the other ram loci. Specifically, ram-4(bx25ts) behaves like the deficiency, mDf9, in its trans-heterozygous interactions with mutations at other ram loci. Unfortunately, these studies were limited to assaying gene interactions between ram alleles that were not necessarily nulls. Putative null alleles of the ram genes have since been obtained and verified and combined-haplo-insufficiency has been observed for some but not all of the ram loci (K.L. Chow, pers. comm.). Specifically, ram-1(wx71), ram-2(bx76), and ram-4(bx48) exhibit non-allelic non-complementation with one another, but not with ram-6(wx66) (K.L. Chow, pers. comm.). Further work has demonstrated that ram-1, ram-2/ram-3 and ram-4 encode cuticular collagens (Tam et al., International C. elegans Meeting 2003, 81; Tam et al., in prep.; Yu and Chow, International C. elegans Meeting 2001, 424; Yu et al., in prep.).
Non-allelic non-complementation has been noted in other processes. In particular, in a screen for suppressors of glp-1, sog-1 alleles were demonstrated to not complement alleles of five other sog loci for glp-1 suppression (Maine and Kimble, 1993). In addition, in a screen for suppressors of rol-3 lethality, srl-2(s2506) was demonstrated to not complement mutations in srl-1 (Barbazuk et al., 1994). Unfortunately, molecular information is currently unavailable for these genes and mutations.
More recently, Chang et al. have reported non-allelic non-complementation between genes required for establishing left-right asymmetry of the ASE chemosensory neurons (Chang et al., 2003). These researchers identified a number of lsy (lim-6 symmetry) mutants that had altered asymmetric expression of ASEL or ASER specific reporter constructs. Mutations in one class were identified as alleles of unc-37 and cog-1. Mutations in this class exhibited ectopic expression of an ASEL specific reporter, gcy-7::gfp, in ASER. These researchers also observed ectopic expression of gcy-7::gfp in ASER in an unc-37(e262)/+;+/cog-1(ot28) trans-heterozygote whereas there is only ASEL expression in either heterozygote alone. unc-37 encodes the C. elegans ortholog of the Groucho transcription co-factor and cog-1 encodes the ortholog of vertebrate Nkx6 type homeobox genes (Palmer et al., 2002; Pflugrad et al., 1997). Biochemical studies have demonstrated that vertebrate COG-1 ortholog interacts with Drosophila Groucho through a conserved engrailed homolog (eh1) domain, thus it is likely that UNC-37 and COG-1 physically interact (Muhr et al., 2001). Other lsy mutants in the same class exhibit non-allelic non-complementation with unc-37 and cog-1; however, these have yet to be characterized (Oliver Hobert pers. comm.).
†In Drosophila, "second-site non-complementation" or SSNC is the predominant title for this interaction, however to avoid confusion with intragenic mutations that modify allelic mutations as in the case of "second-site suppressors", we have opted to stay with the "nonallelic non-complementation" title.
Complementation occurs when two mutations together result in a wild-type phenotype. Non-complementation occurs when two mutations together result in a mutant phenotype. The complementation test is a simple and fundamental assay in genetics used to assign a mutation to a gene. However, if this test does not give a straightforward answer, valuable inferences about gene function and interaction may still be obtained if the situation is properly understood.
This chapter was completed after valuable feedback from members of the Hodgkin Lab, D. Williams, M. Best, K. L. Chow and an anonymous reviewer, all to whom I am very appreciative. I am indebted to O. Hobert, K. L. Chow and J. Kimble for sharing unpublished data. KJY was supported by a Ruth L. Kirchstein NRSA (F32A1050333) from NIH.
Ahmed, S., Maruyama, I.N., Kozma, R., Lee, J., Brenner, S., and Lim, L. (1992). The Caenorhabditis elegans unc-13 gene product is a phospholipid-dependent high-affinity phorbol ester receptor. Biochem. J. 287 (Pt 3), 995–999. Abstract
Alfonso, A., Grundahl, K., Duerr, J.S., Han, H.P., and Rand, J.B. (1993). The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter. Science 261, 617–619. Abstract
Alfonso, A., Grundahl, K., McManus, J.R., Asbury, J.M., and Rand, J.B. (1994). Alternative splicing leads to two cholinergic proteins in Caenorhabditis elegans. J. Mol. Biol. 241, 627–630. Abstract Article
Baird, S.E., and Emmons, S.W. (1990). Properties of a class of genes required for ray morphogenesis in Caenorhabditis elegans. Genetics 126, 335–344. Abstract
Barbazuk, W.B., Johnsen, R.C., and Baillie, D.L. (1994). The generation and genetic analysis of suppressors of lethal mutations in the Caenorhabditis elegans rol-3(V) gene. Genetics 136, 129–143. Abstract
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. Abstract
Chang, S., Johnston, R.J., Jr., and Hobert, O. (2003). A transcriptional regulatory cascade that controls left/right asymmetry in chemosensory neurons of C. elegans. Genes Dev. 17, 2123–2137. Abstract Article
Changeux, J.P., and Edelstein, S.J. (1998). Allosteric receptors after 30 years. Neuron 21, 959–980. Abstract Article
Cox, G.N., Fields, C., Kramer, J.M., Rosenzweig, B., and Hirsh, D. (1989). Sequence comparisons of developmentally regulated collagen genes of Caenorhabditis elegans. Gene 76, 331–344. Abstract Article
Crick, F.H., and Orgel, L.E. (1964). The theory of inter-allelic complementation. J. Mol. Biol. 8, 161–165. Abstract
De Melo, J.V., De Souza, W., and Peixoto, C.A. (2002). Ultrastructural analyses of the Caenorhabditis elegans DR 847 bli-1(n361) mutant which produces abnormal cuticle blisters. J. Submicrosc. Cytol. Pathol. 34, 291–297. Abstract
Ferguson, E.L., and Horvitz, H.R. (1985). Identification and characterization of 22 genes that affect the vulval cell lineages of the nematode Caenorhabditis elegans. Genetics 110, 17–72. Abstract
Francis, R., Barton, M.K., Kimble, J., and Schedl, T. (1995). gld-1, a tumor suppressor gene required for oocyte development in Caenorhabditis elegans. Genetics 139, 579–606. Abstract
Fuller, M.T., Regan, C.L., Green, L.L., Robertson, B., Deuring, R., and Hays, T.S. (1989). Interacting genes identify interacting proteins involved in microtubule function in Drosophila. Cell Motil Cytoskeleton 14, 128–135. Abstract Article
Greenwald, I.S., and Horvitz, H.R. (1980). unc-93(e1500): a behavioral mutant of Caenorhabditis elegans that defines a gene with a wild-type null phenotype. Genetics 96, 147–164. Abstract
Gupta, M.C., Graham, P.L., and Kramer, J.M. (1997). Characterization of alpha1(IV) collagen mutations in Caenorhabditis elegans and the effects of alpha1 and alpha2(IV) mutations on type IV collagen distribution. J. Cell. Biol. 137, 1185–1196. Abstract Article
Hawley, R.S., and Walker, M.Y. (2003). Advanced genetic analysis: finding meaning in a genome (Malden, Massachusetts: Oxford, Blackwell).
Hays, T.S., Deuring, R., Robertson, B., Prout, M., and Fuller, M.T. (1989). Interacting proteins identified by genetic interactions: a missense mutation in alpha-tubulin fails to complement alleles of the testis-specific beta-tubulin gene of Drosophila melanogaster. Mol. Cell Biol. 9, 875–884. Abstract
Hill, R.J., and Sternberg, P.W. (1992). The gene lin-3 encodes an inductive signal for vulval development in C. elegans. Nature 358, 470–476. Abstract Article
Hwang, B.J., and Sternberg, P.W. (2004). A cell-specific enhancer that specifies lin-3 expression in the C. elegans anchor cell for vulval development. Development 131, 143–151. Abstract Article
Jackson, S.M., and Berg, C.A. (1999). Soma-to-germline interactions during Drosophila oogenesis are influenced by dose-sensitive interactions between cut and the genes cappuccino, ovarian tumor and agnostic. Genetics 153, 289–303. Abstract
Johnstone, I.L. (2000). Cuticle collagen genes. Expression in Caenorhabditis elegans. Trends Genet. 16, 21–27. Abstract Article
Jones, A.R., and Schedl, T. (1995). Mutations in gld-1, a female germ cell-specific tumor suppressor gene in Caenorhabditis elegans, affect a conserved domain also found in Src-associated protein Sam68. Genes Dev. 9, 1491–1504. Abstract
Kidd, T., Bland, K.S., and Goodman, C.S. (1999). Slit is the midline repellent for the robo receptor in Drosophila. Cell 96, 785–794. Abstract Article
Killeen, M., Tong, J., Krizus, A., Steven, R., Scott, I., Pawson, T., and Culotti, J. (2002). UNC-5 function requires phosphorylation of cytoplasmic tyrosine 482, but its UNC-40-independent functions also require a region between the ZU-5 and death domains. Dev. Biol. 251, 348–366. Article
Kramer, J.M., and Johnson, J.J. (1993). Analysis of mutations in the sqt-1 and rol-6 collagen genes of Caenorhabditis elegans. Genetics 135, 1035–1045. Abstract
Kramer, J.M., Johnson, J.J., Edgar, R.S., Basch, C., and Roberts, S. (1988). The sqt-1 gene of C. elegans encodes a collagen critical for organismal morphogenesis. Cell 55, 555–565. Abstract Article
Kusch, M., and Edgar, R.S. (1986). Genetic studies of unusual loci that affect body shape of the nematode Caenorhabditis elegans and may code for cuticle structural proteins. Genetics 113, 621–639. Abstract
Liu, J., Tzou, P., Hill, R.J., and Sternberg, P.W. (1999). Structural requirements for the tissue-specific and tissue-general functions of the Caenorhabditis elegans epidermal growth factor LIN-3. Genetics 153, 1257–1269. Abstract
Maine, E.M., and Kimble, J. (1993). Suppressors of glp-1, a gene required for cell communication during development in Caenorhabditis elegans, define a set of interacting genes. Genetics 135, 1011–1022. Abstract
Malone, C.J., Fixsen, W.D., Horvitz, H.R., and Han, M. (1999). UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development 126, 3171–3181. Abstract
Maruyama, I.N., and Brenner, S. (1991). A phorbol ester/diacylglycerol-binding protein encoded by the unc-13 gene of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 88, 5729–5733. Abstract
McKay, J.P., Raizen, D.M., Gottschalk, A., Schafer, W.R., and Avery, L. (2004). eat-2 and eat-18 are required for nicotinic neurotransmission in the Caenorhabditis elegans pharynx. Genetics 166, 161–169. Abstract Article
Meneely, P.M., and Herman, R.K. (1979). Lethals, steriles and deficiencies in a region of the X chromosome of Caenorhabditis elegans. Genetics 92, 99–115. Abstract
Meneely, P.M., and Herman, R.K. (1981). Suppression and function of X-linked lethal and sterile mutations in Caenorhabditis elegans. Genetics 97, 65–84. Abstract
Merz, D.C., Zheng, H., Killeen, M.T., Krizus, A., and Culotti, J.G. (2001). Multiple signaling mechanisms of the UNC-6/netrin receptors UNC-5 and UNC-40/DCC in vivo. Genetics 158, 1071–1080. Abstract
Muhr, J., Andersson, E., Persson, M., Jessell, T.M., and Ericson, J. (2001). Groucho-mediated transcriptional repression establishes progenitor cell pattern and neuronal fate in the ventral neural tube. Cell 104, 861–873. Abstract Article
Myllyharju, J., and Kivirikko, K.I. (2004). Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43. Abstract Article
Novelli, J., Ahmed, S., and Hodgkin, J. (2004). Gene interactions in Caenorhabditis elegans define DPY-31 as a candidate procollagen C-proteinase and SQT-3/ROL-4 as its predicted major target. Genetics 168, 1259–1273. Abstract Article
Page, A.P., and Winter, A.D. (2003). Enzymes involved in the biogenesis of the nematode cuticle. Adv. Parasitol. 53, 85–148. Abstract
Palmer, R.E., Inoue, T., Sherwood, D.R., Jiang, L.I., and Sternberg, P.W. (2002). Caenorhabditis elegans cog-1 locus encodes GTX/Nkx6.1 homeodomain proteins and regulates multiple aspects of reproductive system development. Dev. Biol. 252, 202–213. Abstract Article
Park, E.C., and Horvitz, H.R. (1986). C. elegans unc-105 mutations affect muscle and are suppressed by other mutations that affect muscle. Genetics 113, 853–867. Abstract
Peters, K., McDowall, J., and Rose, A.M. (1991). Mutations in the bli-4 (I) locus of Caenorhabditis elegans disrupt both adult cuticle and early larval development. Genetics 129, 95–102. Abstract
Pflugrad, A., Meir, J.Y., Barnes, T.M., and Miller, D.M., III. (1997). The Groucho-like transcription factor UNC-37 functions with the neural specificity gene unc-4 to govern motor neuron identity in C. elegans. Development 124, 1699–1709. Abstract
Raizen, D.M., Lee, R.Y., and Avery, L. (1995). Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans. Genetics 141, 1365–1382. Abstract
Rancourt, D.E., Tsuzuki, T., and Capecchi, M.R. (1995). Genetic interaction between hoxb-5 and hoxb-6 is revealed by nonallelic noncomplementation. Genes Dev. 9, 108–122. Abstract
Rand, J.B. (1989). Genetic analysis of the cha-1-unc-17 gene complex in Caenorhabditis. Genetics 122, 73–80.
Rand, J.B., and Russell, R.L. (1984). Choline acetyltransferase-deficient mutants of the nematode Caenorhabditis elegans. Genetics 106, 227–248. Abstract
Richmond, J.E., and Broadie, K.S. (2002). The synaptic vesicle cycle: exocytosis and endocytosis in Drosophila and C. elegans. Curr. Opin. Neurobiol 12, 499–507. Abstract Article
Rose, A.M., and Baillie, D.L. (1980). Genetic organization of the region around UNC-15 (I), a gene affecting paramyosin in Caenorhabditis elegans. Genetics 96, 639–648. Abstract
Sibley, M.H., Johnson, J.J., Mello, C.C., and Kramer, J.M. (1993). Genetic identification, sequence, and alternative splicing of the Caenorhabditis elegans alpha 2(IV) collagen gene. J. Cell Biol. 123, 255–264. Abstract Article
Stearns, T., and Botstein, D. (1988). Unlinked noncomplementation: isolation of new conditional-lethal mutations in each of the tubulin genes of Saccharomyces cerevisiae. Genetics 119, 249–260. Abstract
Thacker, C., Peters, K., Srayko, M., and Rose, A.M. (1995). The bli-4 locus of Caenorhabditis elegans encodes structurally distinct kex2/subtilisin-like endoproteases essential for early development and adult morphology. Genes Dev. 9, 956–971. Abstract
Thacker, C., Srayko, M., and Rose, A.M. (2000). Mutational analysis of bli-4/kpc-4 reveals critical residues required for proprotein convertase function in C. elegans. Gene 252, 15–25. Abstract Article
van der Keyl, H., Kim, H., Espey, R., Oke, C.V., and Edwards, M.K. (1994). Caenorhabditis elegans sqt-3 mutants have mutations in the col-1 collagen gene. Dev. Dyn. 201, 86–94. Abstract Article
Wood, W.B. (1988). The nematode Caenorhabditis elegans, (Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory).
Yook, K.J., Proulx, S.R., and Jorgensen, E.M. (2001). Rules of nonallelic noncomplementation at the synapse in Caenorhabditis elegans. Genetics 158, 209–220. Abstract
*Edited by Jonathan Hodgkin and Philip Anderson. Last revised October 05, 2005. Published October 06, 2005. This chapter should be cited as: Yook, K. Complementation (October 06, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.24.1, http://www.wormbook.org.
Copyright: © 2005 Karen Yook. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: karen.yook@bioch.ox.ac.uk
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.