Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Several forward and reverse proteomic approaches are available that can be used to identify interaction partners for a protein of interest. Here we provide methods for identifying interacting partners by the yeast two-hybrid system (a reverse proteomic method) and by tandem immuno-affinity purification of protein complexes combined with mass spectrometry (a forward proteomic method).
The yeast two-hybrid system is a convenient reverse proteomic approach to identify and study protein-protein interactions (Fields and Song, 1989; Vidal and Legrain, 1999; Walhout and Vidal, 2001). The yeast two-hybrid system can be used with a single bait protein of interest. However, large datasets in which many C. elegans proteins were used as baits are now also available (Boulton et al., 2002; Davy et al., 2001; Li et al., 2004; Tewari et al., 2004; Walhout et al., 2002; Walhout et al., 2000).
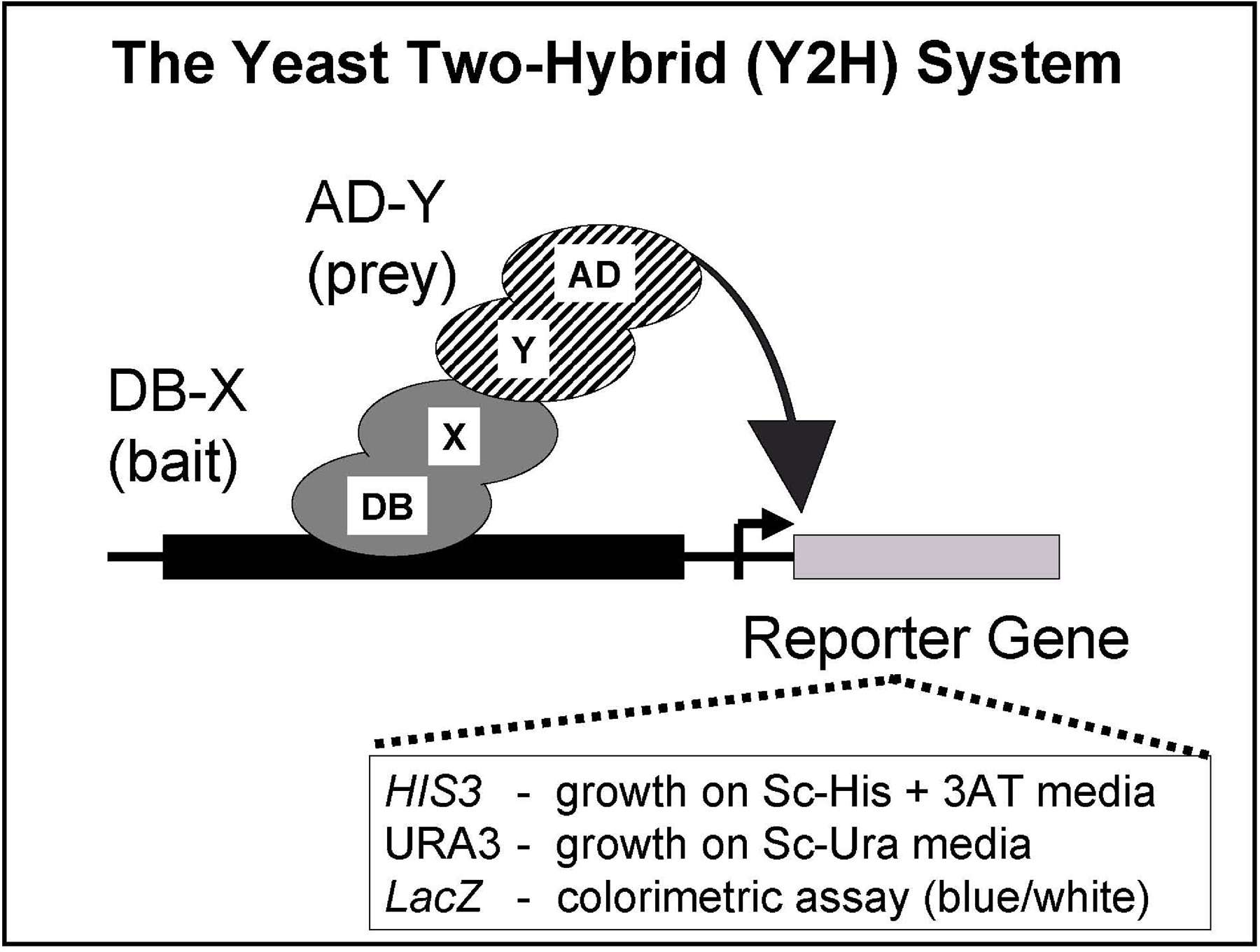
The yeast two-hybrid system is based on the functional reconstitution of an intact transcription factor that activates reporter gene expression (Figure 1). Reporter gene expression can efficiently be selected for in yeast. As indicated by the name, the yeast two-hybrid system utilizes two hybrid proteins. The bait protein (X; Figure 1) is fused to the DNA binding domain (DB; Figure 1) of a transcription factor. The prey protein (Y; Figure 1) is fused to the transcription activation domain (AD; Figure 1) of a transcription factor. In our system, we use the yeast transcription factor Gal4. When two hybrid proteins are co-expressed in yeast and when X and Y can physically interact with each other, a functional transcription factor is reconstituted. This reconstituted transcription factor activates the expression of a set of reporter genes. To minimize the number of false positives identified, we use 3 reporter genes: HIS3, URA3 and lacZ. Expression of these genes is measured by growth on minimal media lacking histidine (for HIS3), or uracil (URA3), or by a colorimetric (“blue-white”) assay.
Performing yeast two-hybrid screens consists of the following steps: 1) creating a DB-X bait, 2) testing self-activation by the DB-X bait, 3) performing the yeast two-hybrid screen and 4) identifying the prey. These steps are described below in more detail. For additional information see Vidal and Legrain (1999) and Walhout and Vidal (2001).
Bait-encoding ORFs can be cloned into a DB-containing vector by regular, restriction-enzyme based cloning or by Gateway cloning. For regular cloning, we use the plasmid pPC97 that has the DNA binding domain of Gal4 immediately upstream of a multiple cloning site (Vidal and Legrain, 1999). We generally prefer to use Gateway cloning because many DB-ORF baits can be created simultaneously and in a standardized and high-throughput manner (Hartley et al., 2000; Walhout et al., 2000; Walhout et al., 2000). After creating the DB-ORF clone(s) of interest, they need to be transformed into the yeast strain MaV103 (Vidal, 1997; Vidal and Legrain, 1999; Walhout and Vidal, 2001). A protocol for high-efficiency yeast transformations is provided below. Because DB-ORF-containing plasmids contain a LEU2 marker gene, transformants need to be selected on plates lacking leucine (Sc-Leu). Before proceeding with the actual yeast two-hybrid screen, bait strains need to be examined for self-activation, i.e. their ability to activate reporter gene expression in the absence of an AD-Y interaction partner (see below).
For Gateway LR cloning the following reagents are required:
An Entry clone from the ORFeome that contains the ORF(s) of interest (Reboul et al., 2003). These can be obtained from Dr. Marc Vidal's laboratory, see http://worfdb.dfci.harvard.edu/.
A Destination vector containing the DNA binding domain of Gal4: (pDESTDB; Walhout et al., 2000). This vector can be obtained from Invitrogen, as part of their Gateway-compatible yeast two-hybrid system (catalog number: 10835-031).
Gateway LR enzyme (Invitrogen, catalog number: 11791–020).
Competent DH5α bacteria with a transformation efficiency of >107 colonies/μgDNA.
LB/ampicillin plates (50 μg/ml).
Protocols for Gateway cloning reactions are described elsewhere (Walhout et al., 2000).
After transformation to create the DB-X bait strain, pick 12 individual yeast colonies and patch them onto a fresh Sc-Leu plate. We patch 5 control strains at the bottom of each plate for comparison (see Walhout and Vidal (2001) for details).
Incubate overnight at 30°C.
Next day, replica plate the patches onto 5 Sc-Leu,-His + 3-amintotriazole (3AT) plates (containing 20, 40, 60, 80 and 100 mM 3AT). 3AT is a competitive inhibitor of the His3 enzyme and only baits that exhibit self-activation will be able to grow in the presence of this compound. After replica plating, the 3AT-containing plates need to be replica cleaned 2 or 3 times, until no yeast is visible (Walhout and Vidal, 2001).
Next day, perform lacZ assay (see below for protocol).
Look at the 3AT plates on days 3, 4 and 5 after replica plating. Bait strains that grow on plates containing 100 mM 3AT cannot be used in yeast two-hybrid experiments. For baits that can be used, use the lowest 3AT concentration at which no growth is observed after 5 days in subsequent yeast two-hybrid screens.
We also perform lacZ assays to examine self-activation (see below). Patches that are blue correspond to self-activating baits.
For yeast two-hybrid screens, the transformation protocol indicated below should be followed. For screens, we use 30 μg of a C. elegans AD-Y cDNA library (Walhout et al., 2000). This library contains cDNAs obtained from worms of all developmental stages, males and dauer larvae and is highly comprehensive (Reboul et al., 2001). The vector in which the cDNAs were cloned (pPC86, Vidal, 1997) contains a TRP1 marker for selection of yeast transformants on media lacking tryptophan. The transformation reactions should be directly plated on Sc-Leu,-His,-Trp + 3AT plates to select for colonies in which a protein-protein interaction between a bait and prey protein takes place.
To estimate the transformation efficiency of the reaction, serial dilutions of one reaction (1/10; 1/100 and 1/1000) should be plated on Sc-Leu,-Trp media (Walhout and Vidal, 2001). After incubating for 3 days at 30°C, the number of colonies should be counted and the total number of transformants calculated. We aim to screen at least 1 million colonies per bait.
Incubate the screen plates for 4–5 days at 30°C.
Pick any growing colonies (that are clearly bigger than background colonies, this varies from bait to bait) and patch with a sterile toothpick on a fresh Sc-Leu,-Trp,-His + 3AT plate. Incubate overnight at 30°C.
Next day, replica plate the positives onto a fresh Sc-Leu,-Trp,-His + 3AT plate, a YEPD plate containing a nitrocellulose filter for lacZ assays (see below) and a Sc-Leu,-Trp,-Ura plate. Replica clean the 3AT containing plate and the Sc-Leu, -Trp,-Ura plate until no yeast is visible. Incubate at 30°C.
Next day, perform the lacZ assay (see below).
Examine growth of potential positives on the other two plates after incubating for 3–5 days. Any growing patches are considered positives. We only consider colonies that score positive for at least 2 reporter genes, e.g., they grow on 3AT-containing media and are blue.
Retest each interaction in fresh bait-containing yeast cells by PCR/Gap-repair (Walhout and Vidal, 2001) to minimize the number of false positives.
Patch the required yeast strain on a YEPD plate and incubate overnight at 30°C.
Next day, inoculate 500 ml liquid YEPD media with the relevant yeast strain to a density of OD600 = ~0.1.
Incubate at 30°C while shaking vigorously until the culture reaches an OD600 between 0.4–0.5.
Harvest the cells by centrifugation for 5 minutes at 1,800 rpm at room temperature.
Decant the medium and wash the cells with 50 ml sterile water.
Resuspend the cells by pipetting up and down. Centrifuge 5min. at 1,800 rpm and RT - decant water.
Wash the cells in 10 ml TE/LiAc (10 mM Tris-HCl, pH 8.0; 1 mM EDTA; 0.1 M lithium acetate), centrifuge as described above.
Resuspend the cells in a volume of TE/LiAc solution corresponding to 10 times the final OD600 value (i.e. for a 500 ml culture with an OD600 = 0.5, 5 ml TE/LiAc solution should be used).
Add 0.1 volume boiled (bring to a boil, and put on ice) salmon sperm DNA (10 mg/ml stock solution; Sigma, catalog number: D9156).
For each tranformation reaction put 50 μl of the cell suspension in a 1.5 ml eppendorf tube.
Add the DNA to be transformed. For screens we use 30 reactions, each with 1μg library DNA.
Add the DNA to be transformed and 300 μl TE/LiAc/PEG solution (made from a 10X stock solutions of TE and LiAc with the remaining volume being polyethylene glycol Mw 3350).
Resuspend carefully by pipetting.
Incubate at least 30 minutes at 30°C.
Heat-shock the reactions for exactly 20 minutes at 42°C.
Quickly centrifuge the tubes in a microcentrifuge, 5 seconds is sufficient.
Remove the supernatant.
Resuspend the cells in 350 μl sterile water and plate on the appropriate selective media (i.e., Sc-Leu,-Trp,-His with the appropriate concentration of 3AT; Walhout and Vidal, 2001).
Yeast should be grown overnight at 30°C on a nitrocellulose filter that is placed on top of a YEPD plate (Walhout and Vidal, 2001). We use filters from Osmonics (catalog number: WP4HY13700).
Next day, place 2 Whatman paper filters (Whatman catalog number: 1454125) in an empty 15 cm Petri dish.
Soak the filters with 6 ml Z-buffer (Walhout and Vidal, 2001) containing 11 μl β-mercaptoethanol and 100 μl 4% X-gal solution (dissolved in dimethylformamide). This should be done in a chemical hood.
Take the nitrocellulose filter containing the yeast with a pair of tweezers and quick-freeze for 10 seconds in liquid nitrogen. Thaw while holding with the tweezers.
Carefully place the nitrocellulose on top of the Whatman filters, with the yeast patches facing up.
Incubate overnight at 37°C.
Blue yeast patches correspond to self-activating baits (when only DB-X is used), or to potential positives (when DB-X is screened against an AD-Y cDNA library).
The ORFs encoding interacting preys can be obtained from yeast by colony PCR using AD and Term primers. For a protocol see: Walhout and Vidal (2001). PCR products can be used in PCR/Gap-repair to retest potential interactions. PCR products corresponding to retesting positives should be purified to remove the primers. Subsequently, PCR products can be sequenced using the AD primer to obtain an IST (interaction sequence tag). ISTs can be blasted against Wormpep in Wormbase (http://www.wormbase.org/db/searches/blat) to identify the prey.
Multi-protein complexes are the functional units of many cellular processes. Defining the components of these complexes, how they associate and their intrinsic biochemical activities provides a wealth of information about the context in which proteins operate (Deshaies et al., 2002; Gavin et al., 2002; Ho et al., 2002; Rigaut et al., 1999; Shevchenko et al., 2002). Here we describe a standardized method for purification of protein complexes from C. elegans extracts by tandem immuno-affinity (Polanowska et al., 2004). This method describes the following steps: 1). Creating the transgene for expression of an epitope tagged form of your protein of interest (P.O.I.), 2). Growth and harvesting the transgenic line, 3). Generating lysates, 4) Preparing antibody coupled beads, 5) Tandem immuno-affinity purification of protein complexes, and 6) Preparation of the sample for mass-spectrometry (MS).
We have constructed pSB_GW::TAG, a Gateway destination vector that can be used to generate transgenic animals that express any P.O.I. fused at the C-terminus to HA_8xHis_TEV_Myc epitopes, enabling associated protein complexes to be purified from whole worm or embryo extracts by tandem immuno-affinity (Figure 2A; Polanowska et al., 2004). See (Walhout et al., 2000) for details on Gateway cloning. The promoter and coding region of a gene of interest (G.O.I.) are cloned into the Gateway entry vector, sequenced, and then transferred by Gateway LR cloning into pSB_GW::TAG as previously described (Polanowska et al., 2004; Figure 2A). Stable transgenic animals generated by microparticle bombardment (Praitis et al., 2001) of unc-119(ed3) mutants with pSB_G.O.I::TAG are then tested for transgene expression by western blotting using antibodies against either 1) the HA epitope (MAb12CA5), 2) 8xHis (anti-His antibody, Amersham BioSciences), or 3) the Myc epitope (MAb9E10).
|
Figure 2. Protein complex purification from C. elegans. (A) Gateway recombination is used to clone the promoter and coding region for a gene of interest (G.O.I) into p221 using the BP reaction, and from p221 into pSB_GW::TAG using the LR reaction. An example of this is shown on the right in which universal primers have been used to PCR amplify the promotor and coding region (P+C) for Y41E3.9 from pSB_Y41E3.9::TAG (B) Microparticle bombardment is used to integrate the construct into unc-119(ed3) mutants at low copy to generate a transgenic line expressing the protein of interest (P.O.I.) fused to C-terminal epitope tags. The transgenic line is grown to high density in a BioFlo5000® fermenter, harvested and extracts generated for purification purposes(C). Tandem immuno-affinity chromatography over protein-A-MAb12CA5 and then protein-A-MAb9E10 is used to purify the P.O.I.-associated protein complex, before complex components are identified by mass-spectrometry. An example on the right shows a silver stained 4–12% gradient gel comparing the mock purification after (1.) HA and (2.) HA + Myc to the (3.) Y41E3.9 associated protein complex after HA + MYC immunoaffinity.
We have optimized large-scale production of transgenic lines in BioFlo5000® fermenters (New Brunswick Scientific) or alternatively, smaller scale cultures grown in S-basal may yield sufficient material for purification purposes. See elsewhere for details (Polanowska et al., 2004). The weight of worms required for purifying a given protein complex to sufficient levels for protein identification by MS will depend on the level of expression from the genes promotor and must be determined for each transgene. The complexes we have studied so far function in the DNA damage response and are expressed at low levels in all proliferating cells, but are undetectable in somatic tissues. For all complexes (n=5) studied approximately 50g of biomass was required to obtained sufficient protein for MS.
To prepare the worms for lysis:
Wash the worms twice in 4x the pellet volume of cold 0.1M NaCl.
Centrifugate at 4,000 rpm in 30% sucrose at 4°C for 5 minutes to remove residual bacteria and dead worms (Figure 2B).
Collect the live worms from the top of the sucrose solution.
Wash a third time with 4x the pellet volume of cold 0.1M NaCl.
Resuspend the pellet in twice the worm pellet volume of CSK lysis buffer [100mM Pipes (pH 6.0), 100mM NaCl, 3mM MgCl2, 1mM EGTA, 1mM DTT, 1mM PMSF, 0.3M sucrose, 0.5% triton X 100, complete (Roche) protease inhibitor tablets, and phosphatase inhibitors: 2mM β-glycerophosphate, 5mM NaF, 1mM Na3VO4, 0.1 μM okadaic acid].
Freeze down in 50 ml aliquots on dry ice and store at -80°C.
To generate whole worm extracts perform the following:
Rapidly thaw the aliquot(s) in a 37°C water bath.
Re-freeze on dry ice then re-thaw as before.
Dounce the suspension 6 times in a homogenizer to break up the material.
Clarify the mixture by centrifugation at 4,500 rpm and collect the soluble supernatant fraction (S).
To extract the chromatin fraction (C), resuspend the pellet in half the pellet volume of CSK.
Add micrococcal nuclease (Roche) to a final concentration of 3U/μl.
Incubate at 20°C with gentle shaking for 30 minutes.
Stop the reaction by adding EDTA and EGTA to a final concentration of 5mM.
Collection the chromatin fraction supernatant by high-speed centrifugation at 40,000 rpm.
Lysates can also be prepared from embryos:
Resuspend the embryo pellet in twice the pellet volume of homogenization buffer (50 mM HEPES at pH 7.6, 200 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.2% Triton X-100, 5% glycerol, complete (Roche) protease inhibitor tablets, and phosphatase inhibitors: 2mM β-glycerophosphate, 5mM NaF, 1mM Na3VO4, 0.1 μM okadaic acid).
Sonicate on ice until the mixture has lost its viscosity.
Centrifuge at 5,000g at 4°C for 5 min and collect the soluble extract (S).
The chromatin bound fraction (C) is prepared as above.
Incubate 1 ml sepharose protein-A beads (Pharmacia) at 20°C with mouse ascite (2 mg of MAb12CA5(HA) and MAb9E10 (Myc) antibodies).
Wash the beads with 10 volumes of 0.2 M sodium borate (pH 9.0).
Add dimethylpimelimidate dihydrochloride (Sigma) to a final concentration of 2 mM to couple the antibody to the beads.
Incubate for 30 minutes at 20°C.
Wash the beads with 5 volumes of 0.1 M Tris-HCl (pH 8.0) and 10 volumes of PBS to stop the coupling reaction.
The coupled beads are stored at 4°C in two volumes of PBS in 0.5% azide. Prior to purification, the beads should be washed in B0.1 buffer [50 mM Tris (pH 7.3), 0.1 M KCl, 1 mM EDTA, 10% glycerol].
The first immuno-affinity step:
Batch absorb at 4°C for 6 hours with 60 μl HA coupled protein-A beads for each ml of extract (Figure 1C).
Transfer the beads to a BioRad column.
Wash three times with 5 volumes of B0.1 buffer [supplemented with 1 mM DTT, 1mM PMSF, 2mM β-glycerophosphate, 5 mM NaF, 1 mM Na3VO4, 1 μM okadaic acid and complete protease inhibitor tablets].
Elute the bound complex by resuspending the beads in one volume of B0.1 buffer containing 5 μg/ml of double HA peptide.
Incubate at 4°C for 2 hrs.
Using the same conditions as above, the HA eluate is subjected to a second immuno-affinity step using Myc coupled protein-A beads (Figure 2C). Elution from Myc-beads is inefficient using Myc-peptide so recombinant TEV protease (Invitrogen) is used to cleave off the Myc epitope. The cleavage reaction is performed at 20°C for 4 hours at a concentration of 1 μl TEV per 50 μl of HA eluate. The HA-Myc eluate can be further concentrated by binding to Ni-agarose beads followed by elution using a 20–500mM imidazole gradient in B0.1 buffer.
To minimize contamination with keratins replace gloves frequently. Also, resolve purified protein complexes on Nu-PAGE 4–12% pre-poured gels (Invitrogen) and stain with either Coomassie stain or silver stain (SilverQuest, Invitrogen). Bands are excised from the gel and prepared for MS analysis as described previously (Shevchenko et al., 1996). Coomassie stainable levels of protein should be sufficient for protein identification by MS but it is important to discuss sample preparation with your MS lab/facility as they may have specific requirements. To obtain a protein ID, search the databases at PepMAPPER and MASCOT with the peak masses obtained from MS. Note that mock tandem immuno-affinity purification from extracts made from an untagged C. elegans strain occasionally gives two bands, one of which is HSP-70 (C12C8.1; data not shown).
SB would like to thank Biotechiques for permitting the reproducion of some of the work published in Polanowska et al, 2004, Biotechniques 36:778–780, 782. AJMW is funded by NCI (R33 CA097516–02), the Worcester Foundation and a Diabetes Endocrinology Research Center Grant 5 P30 DK32520. SB is funded by Breast Cancer Campaign (GA3221) and Cancer Research UK.
Boulton, S.J., Gartner, A., Reboul, J., Vaglio, P., Dyson, N., Hill, D.E., and Vidal, M. (2002). Combined functional genomic maps of the C. elegans DNA damage response. Science 295, 127–131. Abstract Article
Davy, A., Bello, P., Thierry-Mieg, N., Vaglio, P., Hitti, J., Doucette-Stamm, L., Thierry-Mieg, D., Reboul, J., Boulton, S., Walhout, A.J., et al. (2001). A protein-protein interaction map of the Caenorhabditis elegans 26S proteasome. EMBO Rep. 2, 821–828. Abstract Article
Deshaies, R.J., Seol, J.H., McDonald, W.H., Cope, G., Lyapina, S., Shevchenko, A., Verma, R., and Yates, J.R., III (2002). Charting the protein complexome in yeast by mass spectrometry. Mol. Cell Proteomics 1, 3–10. Abstract Article
Fields, S., and Song, O. (1989). A novel genetic system to detect protein-protein interactions. Nature 340, 245–246. Abstract Article
Gavin, A.C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J.M., Michon, A.M., Cruciat, C.M., et al. (2002). Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147. Abstract Article
Hartley, J.L., Temple, G.F., and Brasch, M.A. (2000). DNA cloning using in vitro site-specific recombination. Genome Res. 10, 1788–1795. Abstract Article
Ho, Y., Gruhler, A., Heilbut, A., Bader, G.D., Moore, L., Adams, S.L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. (2002). Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183. Abstract Article
Li, S., Armstrong, C.M., Bertin, N., Ge, H., Milstein, S., Boxem, M., Vidalain, P.O., Han, J.D., Chesneau, A., Hao, T., et al. (2004). A map of the interactome network of the metazoan C. elegans. Science 303, 540–543. Abstract Article
Polanowska, J., Martin, J.S., Fisher, R., Scopa, T., Rae, I., and Boulton, S.J. (2004). Tandem immunoaffinity purification of protein complexes from Caenorhabditis elegans. Biotechniques 36, 778–780, 782. Abstract
Praitis, V., Casey, E., Collar, D., and Austin, J. (2001). Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157, 1217–1226. Abstract
Reboul, J., Vaglio, P., Rual, J.F., Lamesch, P., Martinez, M., Armstrong, C.M., Li, S., Jacotot, L., Bertin, N., Janky, R., Moore, T., Hudson, J.R., Jr., Hartley, J.L., Brasch, M.A., Vandenhaute, J., Boulton, S., Endress, G.A., Jenna, S., Chevet, E., Papasotiropoulos, V., Tolias, P.P., Ptacek, J., Snyder, M., Huang, R., Chance, M.R., Lee, H., Doucette-Stamm, L., Hill, D.E., and Vidal, M. (2003). C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat. Genet. 34, 35–41. Abstract Article
Reboul, J., Vaglio, P., Tzellas, N., Thierry-Mieg, N., Moore, T., Jackson, C., Shin-i, T., Kohara, Y., Thierry-Mieg, D., Thierry-Mieg, J., et al. (2001). Open-reading-frame sequence tags (OSTs) support the existence of at least 17, 300 genes in C. elegans. Nat. Genet. 27, 332–336. Abstract Article
Rigaut, G., Shevchenko, A., Rutz, B., Wilm, M., Mann, M., and Seraphin, B. (1999). A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17, 1030–1032. Abstract Article
Shevchenko, A., Schaft, D., Roguev, A., Pijnappel, W.W., and Stewart, A.F. (2002). Deciphering protein complexes and protein interaction networks by tandem affinity purification and mass spectrometry: analytical perspective. Mol. Cell Proteomics 1, 204–212. Abstract Article
Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858. Abstract Article
Tewari, M., Hu, P.J., Ahn, J.S., Ayivi-Guedehoussou, N., Vidalain, P.O., Li, S., Milstein, S., Armstrong, C.M., Boxem, M., Butler, M.D., et al. (2004). Systematic interactome mapping and genetic perturbation analysis of a C. elegans TGF-beta signaling network. Mol. Cell. 13, 469–482. Abstract Article
Vidal, M. (1997). The reverse two-hybrid system. In: The Yeast Two-Hybrid System, P. Bartels and S. Fields, Eds. Oxford University Press: New-York, p. 109–147.
Vidal, M., and Legrain, P. (1999). Yeast forward and reverse ‘n’-hybrid systems. Nucleic Acids Res. 27, 919–929. Abstract Article
Walhout, A.J., Boulton, S.J., and Vidal, M. (2000). Yeast two-hybrid systems and protein interaction mapping projects for yeast and worm. Yeast 17, 88–94. Abstract
Walhout, A.J., Reboul, J., Shtanko, O., Bertin, N., Vaglio, P., Ge, H., Lee, H., Doucette-Stamm, L., Gunsalus, K.C., Schetter, A.J., et al. (2002). Integrating interactome, phenome, and transcriptome mapping data for the C. elegans germline. Curr. Biol. 12, 1952–1958. Abstract Article
Walhout, A.J., Sordella, R., Lu, X., Hartley, J.L., Temple, G.F., Brasch, M.A., Thierry-Mieg, N., and Vidal, M. (2000). Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 287, 116–122. Abstract Article
Walhout, A.J., Temple, G.F., Brasch, M.A., Hartley, J.L., Lorson, M.A., van den Heuvel, S., and Vidal, M. (2000). GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 328, 575–592. Abstract
*Edited by Victor Ambros. Last revised April 11, 2005. Published February 13, 2006. This chapter should be cited as: Walhout, A.J.M. and Boulton, S.J. Biochemistry and molecular biology (February 13, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.86.1, http://www.wormbook.org.
Copyright: © 2006 Albertha J. M. Walhout and Simon J. Boulton. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: marian.walhout@umassmed.edu or simon.boulton@cancer.org.uk
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.