Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
The integrity of the genome is essential to the health of the individual and to the reproductive success of a species. Transmission of genetic information is in a selective balance between two opposing forces, the maintenance of genetic stability versus elimination of mutational change and loss of evolutionary potential. Caenorhabditis elegans provides many advantages for the study of DNA surveillance and repair in a multicellular organism. Several genes have been identified by mutagenesis and RNA interference that affect DNA damage checkpoint and repair functions. Many of these DNA damage response genes also play essential roles in DNA replication, cell cycle control, development, meiosis and mitosis. To date, no obvious DNA damage-induced checkpoint has been described in C. elegans somatic cells. In contrast, the DNA damage response in the germ line is characterized by two spatially separate checkpoints; mitotic germ nuclei proliferation arrest and apoptosis of damaged meiotic nuclei. Both of these responses are regulated by checkpoint genes including mrt-2, hus-1, rad-5 and cep-1, the C. elegans ortholog of the human tumour suppressor p53. The germ line DNA damage checkpoints in C. elegans provide an excellent model in which to study the genes required to maintain genomic stability and to test compounds which might have tumor suppressing properties. In addition to single gene studies, integration of data from high-throughput screens has identified genes not previous implicated in the DNA damage response and elucidated novel connections between the different repair pathways. Most of the genes involved are conserved between worms and humans, and in humans, are associated with either oncogenesis or tumor-suppression. Thus, studies of the physical and functional interactions of the components of the repair pathways in C. elegans will provide information about human repair disorders and cancer predisposition.
The integrity of the genome is essential to the health of the individual and to the reproductive success of a species. Faithful transmission of genetic information is in a selective balance between genetic stability and mutational change as a resource for evolutionary potential. In order to maintain genome fidelity the coordinated action of surveillance and repair pathways is required. Much of our knowledge about repair has come from studies in bacteria, yeast and mammals, including human cell lines (reviewed in Sancar et al., 2004; Krogh and Symington, 2004), and it is increasingly evident that most repair functions are highly conserved. Caenorhabditis elegans provides an experimental model in which the repair processes required for both genomic stability and mutational change can be studied. In this Chapter, we present an overview of DNA damage response research using Caenorhabditis elegans. Table 1 is a summary of some DNA damage response genes identified in the C. elegans genome along with mutant phenotypes where known. Included in the Table are components of the pathways for nucleotide excision repair, mismatch repair, DNA damage checkpoint, non-homologous end joining, homologous recombination repair, and chromosomal structure surveillance.
Table 1. List of repair genes and mutant phenotypes
| Pathway | Gene | Cosmid | Mutant phenotype | Reference |
|---|---|---|---|---|
| Uncloned rad mutants | rad-1 | – | UV and gamma sensitive | Hartman and Herman, 1982 |
| rad-2 | – | UV and gamma sensitive | Hartman and Herman, 1982 | |
| rad-3 | – | UV sensitive, reduced brood size | Hartman and Herman, 1982 | |
| rad-4 | – | UV sensitive, cold sensitive, suppressor of ndj | Hartman and Herman, 1982 | |
| rad-6 | – | UV sensitive, Dpy, reduced brood size | Hartman and Herman, 1982 | |
| rad-7 | – | UV sensitive, reduced brood size | Hartman and Herman, 1982 | |
| rad-8 | – | UV sensitive, sick, reduced brood size | Hartman and Herman, 1982 | |
| rad-9 | – | UV sensitive, sick, reduced brood size | Hartman and Herman, 1982 | |
| Nucleotide excision repair | xpa-1 | K07G5.2 | UV sensitive | Park et al., 2002 |
| XPB | Y66D12A.15 | – | – | |
| XPC | Y76B12C.2 | – | – | |
| XPD | Y50D7A.2 | – | – | |
| XPE | M18.5 | Let/Gro | Kamath et al., 2003 | |
| XPF | C47D12.8 | UV sensitive | Park et al., 2004 | |
| XPG | F57B10.6 | Emb | Piano et al., 2002 | |
| ERCC1 | F10G8.7 | – | – | |
| csb-1 | F53H4.1 | UV sensitive | Lee et al., 2002 | |
| Mismatch repair | msh-6 | Y47G6A.11 | Somatic instability and Mut | Tijsterman et al., 2002 |
| msh-2 | H26D21.2 | Microsatellite instability, Mut, reduced DD apoptosis | Degtyareva et al., 2002 | |
| mlh-1 | T28A8.7 | – | Pothof et al., 2003 | |
| pms-2 | H12C20.2 | – | Pothof et al., 2003 | |
| DNA damage checkpoint | mrt-2 | Y41C4A.14 | Mrt, Rad, checkpoint defective | Ahmed and Hodgkin, 2000 |
| hus-1 | H26D21.1 | Mrt, Rad, checkpoint defective | Hofmann et al., 2002 | |
| rad-5/clk-2 | C07H6.6 | Rad, checkpoint defective | Ahmed et al., 2001 | |
| cep-1/p53 | F52B5.5 | Rad, checkpoint defective | Derry et al., 2001; Schumacher et al., 2001 | |
| egl-1 | F23B12.9 | – | Hofmann et al., 2002 | |
| ape-1 | F46F3.4 | Enhanced cep-1-dependent apoptosis | Deng et al., 2004 | |
| abl-1 | M79.1 | Enhanced radiation-induced apoptosis | Deng et al., 2004 | |
| brc-1 | C36A4.8 | Him and increased germ cell apoptosis | Boulton et al., 2004 | |
| brd-1 | K04C2.4 | Him and increased germ cell apoptosis | Boulton et al., 2004 | |
| pme-5 | ZK1005.1 | – | – | |
| kin-20 | F46F2.2 | Rad | – | |
| hpr-9 | Y39A1A.23 | Rad | Boulton et al., 2002 | |
| hpr-17 | F32A11.2 W | Rad | Stergiou and Hengartner, 2004 | |
| chk-1 | Y39H10A.7 | – | – | |
| chk-2 | Y60A3A.12 | Emb Let, Him, absent chiasma in meiosis | MacQueen and Villeneuve, 2001 | |
| Non-homologous end joining | cku-70 | Y47D3A.4 | – | – |
| cku-80 | R07E5.8 | – | Boulton et al., 2002 | |
| lig-4 | C07H6.1 | – | Boulton et al., 2002 | |
| Homologous recombination | atm-1 | Y48G1BL.F | Radiation sensitve, checkpoint defective | Stergiou and Hengartner, 2004; Boulton et al., 2002 |
| atl-1 | T06E4.3 | Embryonic lethality, Him, Rad | Aoki et al., 2000 | |
| rad-54 | W06D4.6 | Rad | Boulton et al., 2002 | |
| rad-50 | T04H1.4 | Rad | – | |
| mre-11 | ZC302.1 | Germline mortality, Him, Emb | Chin and Villeneuve, 2001 | |
| rad-51 | Y43C5A.6 | Embryonic lethality, Him, Rad | Takanami et al., 1998 | |
| top-3 | Y56A3A.27 | – | Wicky et al., 2004 | |
| dna-2 | F43G6.1 | Rad | – | |
| Chromosome structure | coh-2 | F10G7.4 | Late embryo or larval arrest or HIM | – |
| scc-3 | F18E2.3 | Embryonic lethal; cell division defects | – | |
| rec-8 | W02A2.6p | Embryonic lethal; aneuploidy | – | |
| him-1/smc-1 | F28B3.7 | UV sensitive, high incidence of males, Emb | Hartman and Herman, 1982 | |
| him-3 | ZK381.1 | High incidence males, implicated in sister repair | – | |
| Cytokinesis checkpoint | mdf-1 | C50F4.11 | Fail to arrest metaphase to anaphase | – |
| mdf-2 | Y69A2AR.30 | Fail to arrest metaphase to anaphase | – | |
| san-1 | ZC328.4 | Fail to arrest cell cycle during anoxia | – | |
| fzy-1 | ZK177.6 | Embryonic lethal, defective anaphase | – | |
| ify-1/securin | C27A2.3 | Embryonic lethal, defective anaphase | – | |
| sep-1 | Y47G6A.12 | Prevents timely disjunction and segregation | – | |
| ubc-1 | C35B1.1 | – | – | |
| air-2 | B0207.4 | Embryonic lethal; cell division defects | – | |
| Helicases | dog-1 | F33H2.1 | Deletion of G tracts, mutator | Cheung et al., 2002 |
| wrn-1 | F18C5.2 | Abnornmal checkpoint response, premature aging | Lee et al., 2004 | |
| him-6 | T04A11.6 | Embryonic lethality, Him | Wicky et al., 2004 |
In addition to those listed in Table 1 are those genes involved in base excision repair, DNA glycosylation, the rad-6 pathway, trans-lesion bypass, and those encoding editing and processing nucleases.
In C. elegans, the first study of repair function was the identification by Hartman and Herman (1982) of nine Rad (radiation-sensitive) mutants, which were hypersensitive to ultraviolet light during embryogenesis. UV irradiation of the Rad mutants results in a decrease in viability. Many of these Rad mutants exhibit phenotypes in addition to their UV sensitivity suggesting that the Rad genes are involved in other biological processes in addition to their roles in the UV-induced DNA damage response (Table 1). More recently, researchers have used reverse genetic techniques such as RNA interference and PCR-mediated gene knockouts to investigate genes involved in different DNA damage response pathways such as nucleotide excision repair (Park et al., 2002; Park et al., 2004; Lee et al., 2002).
The UV sensitivity of C. elegans varies depending on the developmental stage of the worm (Hartman, 1984; Hartman, 1984) with wild-type animals being most sensitive to UV radiation during early embryogenesis. The UV hypersensitivity of the early stage embryo might be due in part to the rapid cell proliferation and the lack of obvious DNA damage-induced checkpoints; DNA replication progresses even after exposure to large fluences of UV radiation (Jones and Hartman, 1996).
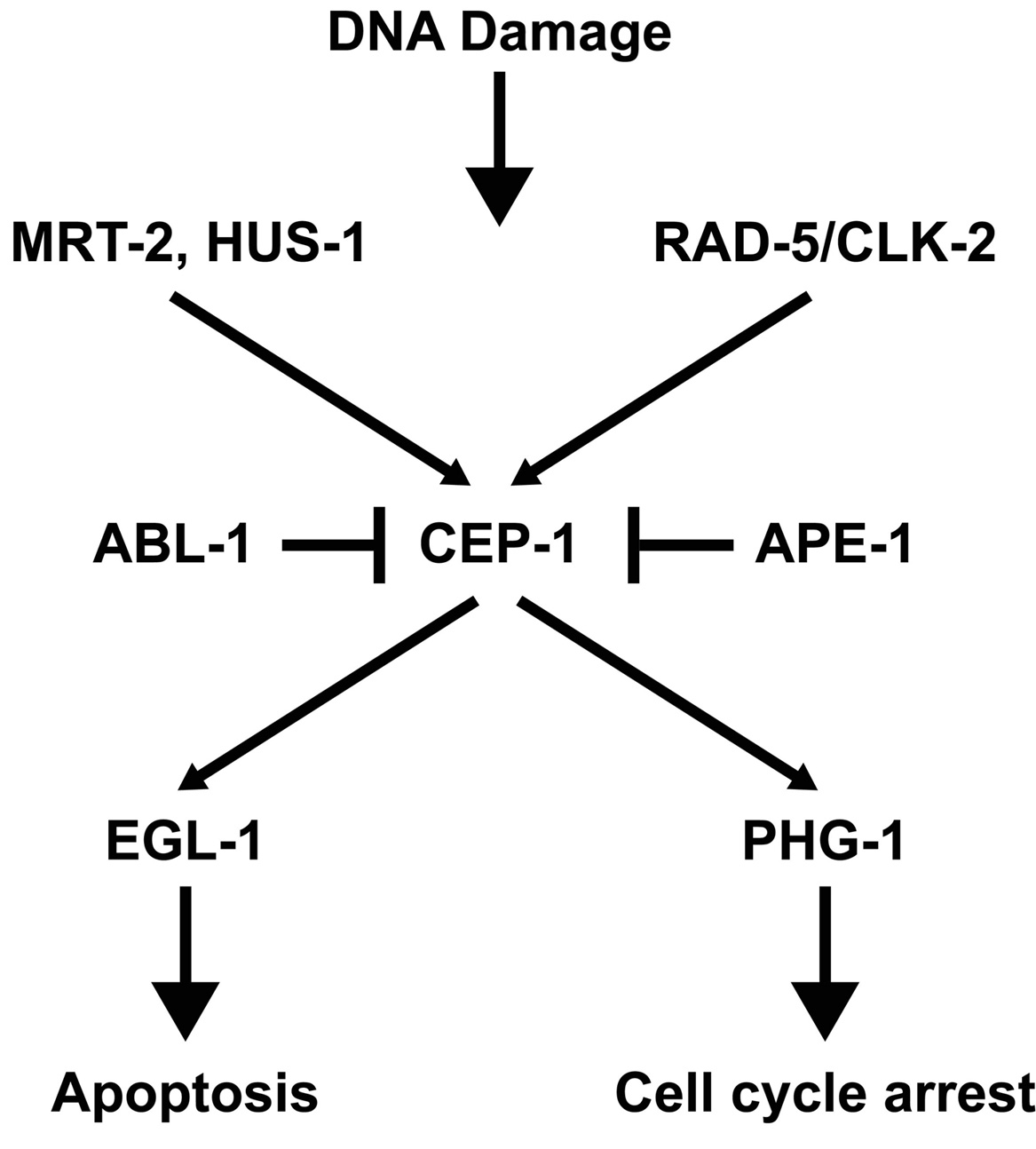
In contrast to the developing embryo, activation of the checkpoint in the germ line results in obvious morphological changes that can be monitored by microscopy of live animals. (Gartner et al., 2000; Stergiou and Hengartner, 2004). In response to DNA damage, mitotic germline nuclei arrest proliferation, presumably to allow time for DNA repair. In the meiotic region of the germ line, cells with DNA damage are removed by apoptosis before oogenesis. DNA damage-induced apoptosis occurs in addition to physiological programmed germ cell death, which is hypothesized to maintain germline homeostasis. DNA damage-induced germ cell death requires the conserved apoptotic machinery (Gartner et al., 2000). However, the proapoptotic gene egl-1, which is required for somatic programmed cell death but not physiological germ cell death, is not absolutely required for DNA damage-induced germ cell apoptosis (Gartner et al., 2000) even though there is a cep-1-dependent increase in egl-1 transcription after genotoxic stress (Hofmann et al., 2002). The specific genes involved in the germline DNA damage-induced checkpoints are shown in Figure 1 and are discussed below.
An exciting finding was the identification of mrt-2, a gene responsible for germline immortality (Ahmed and Hodgkin, 2000). mrt-2 mutants exhibit progressive telomere shortening and accumulate end-to-end chromosome fusions. In addition, mrt-2 mutants are defective in DNA damage-induced mitotic arrest and apoptosis in the germline (Gartner et al., 2000). mrt-2 encodes a homologue of a checkpoint gene required to sense DNA damage in yeast (S. pombe rad1/S. cerevisiae rad17). In addition to mrt-2, function of the DNA damage checkpoint involves the gene hus-1, which when mutated results in a phenotype similar to mrt-2 (Hofmann et al., 2002). Another checkpoint gene is rad-5 (Gartner et al., 2000). Cloning of the rad-5 gene (Ahmed et al., 2001) lead to the surprising result that rad-5(mn159) (from Hartman and Herman, 1982) is allelic with clk-2(qm37), a mutant implicated in regulation of biological rhythms and life-span (Benard et al., 2001; Lim et al., 2001). rad-5/clk-2 regulates the S phase replication checkpoint and is homologous to S. cerevisiae Tel2, which has an essential function in yeast for regulating telomere length. Overexpression of human CLK2 in C. elegans made the cell hypersensitive to apoptosis triggered by oxidative stress or DNA replication block and gradually increased telomere length suggesting that human CLK2 may have a role in maintaining telomere length (Jiang et al., 2003). However, the role of rad-5/clk-2 in the regulation of telomere length remains unclear. Studies investigating the effect of rad-5/clk-2 mutations on telomere length have been contradictory (Lim et al., 2001; Benard et al., 2001; Ahmed et al., 2001). Ahmed and coworkers (2001) suggest that the differences observed in the three studies could be explained by the fact that telomere length varies considerably between C. elegans strains and that telomere length can fluctuate even within isogenic lines. Cheung et al. (2004) used single telomere length analysis (STELA) and reported strain specific differences in wild type as well as mutant strains, such as the telomerase deficient trt-1 (Cheung et al. in press).
p53 is a key regulator of the DNA damage-induced checkpoint in mammals (reviewed in Sancar et al., 2004). cep-1, the C. elegans homolog of p53, is required for DNA damage-induced apoptosis in the C. elegans germ line, but not for programmed cell death occurring during worm development nor physiological (radiation-independent) germ cell death (Schumacher et al., 2001; Derry et al., 2001). Despite the differences in the three-dimensional structure of the DNA binding domain between CEP-1 and human p53 (Huyen et al., 2004) its role in the DNA damage checkpoint appears to be conserved. Furthermore, CEP-1 can induce apoptosis in mammalian cells and this induction can be inhibited by iASPP, an evolutionarily conserved inhibitor of p53 (Bergamaschi et al., 2003).
Several genes have been identified that either regulate cep-1 activity or are regulated by cep-1. The C.elegans iASPP ortholog, ape-1 (apoptotic enhancer) is a conserved inhibitor of cep-1. ape-1(RNAi) results in an increase in cep-1-mediated apoptosis (Bergamaschi et al., 2003). Lettre et al. (2004) also identified ape-1 along with several other genes in a genome-wide RNAi screen for genes that when knocked out resulted in an increase in germ cell death. Many of the genes identified in this screen required cep-1 activity for the increase in germ cell death. Deng et al. (2004) studied the antagonistic effects of abl-1, a homolog of the conserved nonreceptor tyrosine kinase c-Abl, on cep-1-mediated apoptosis. Deletion of abl-1 results in increased radiation-induced apoptosis, but not ethylnitrosourea-induced apoptosis. Thus, ABL-1 can distinguish proapoptotic signals triggered by two different DNA-damaging agents. In addition, treatment of C. elegans with c-Abl inhibitors results in a phenotype similar to the abl-1 mutation, demonstrating the utility of C. elegans as a model to screen for potential anticancer drugs.
In contrast to the Drosophila p53 ortholog, cep-1 has also been shown to regulate DNA damage-induced mitotic germ cell arrest (B. Derry, personal communication). Furthermore, cep-1-mediated germline mitotic arrest is dependent on phg-1, a C. elegans homolog of the human growth arrest gene gas1. phg-1 is required for cep-1-mediated mitotic arrest but not for cep-1-mediated apoptosis indicating that cep-1-mediated mitotic arrest and apoptosis can be separated (B. Derry, personal communication).
Components of the mismatch repair pathway were originally identified in bacteria as mutation-prone strains. In humans, mismatch repair deficiency predisposes to hereditary nonpolyposis colon cancer (HNPCC) and involves the proteins MSH2, MSH6, MLH1, and PMS2. C. elegans has orthologs to all four of these genes. Utilizing a system to screen for repeat sensitivity, Tijsterman et al. (2002) demonstrated that RNAi of msh-2, msh-6, mlh-1 and pms-2 results in a mutator phenotype. In this screening system, a DNA repeat puts a heat-shock promoter-driven lacZ transgene reporter gene out of frame. Mutations that result in a frameshift would result in lacZ expression. In msh-2, msh-6, mlh-1 and pms-2-deficient animals, in-frame lacZ+ patches were observed as a result of somatic repeat instability (Tijsterman et al., 2002; Pothof et al., 2003). Degtyareva et al., (2002) used an msh-2 mutant to demonstrate elevated levels of microsatellite instability. In this study, the phenotype of msh-2 mutants was similar to wild-type worms with regard to lifespan and meiotic chromosome segregation, but msh-2 animals had somewhat reduced fertility. In addition, the mutant worms had reduced DNA damage-induced germ-line apoptosis after genotoxic stress, linking a functional component of the mismatch repair pathway to the DNA damage checkpoint in C. elegans. This result is similar to that observed in mammalian mismatch repair mutants (reviewed in Buermeyer et al., 1999).
Not all mismatch repair homologs have a role in DNA repair. Similar to the situation in budding yeast, the C. elegans MutS family homologs, him-14/msh-4 and msh-5 have no apparent role in repair, but have a specialized role in crossing over during meiosis.
Double-strand breaks (DSBs) occur in response to environmental insult, such as ionizing radiation or genotoxic chemicals, or cellular sources such as oxidative damage or replication blocks in the DNA. In addition to their unwanted creation by DNA damaging events or substances, DSBs are created biologically by SPO-11 as the highly regulated initiation of meiotic recombination. The processing of both categories of DSBs utilizes many of the same proteins and mechanisms. Central to DSB repair is the RecA homolog, Rad51, which mediates functions involving strand invasion during recombination (Shinohara and Ogawa, 1995). RAD-51 localization to DNA requires the products of the recombination genes mre-11 and chk-2 (Rinaldo et al., 2002; Alpi et al., 2003). rad-51 and mre-11 are required for both meiotic recombination and DNA repair (Takanami et al., 2000; Chin and Villeneuve, 2001; Takanami et al., 2003). It is unclear whether the checkpoint kinase, chk-2 functions during DNA repair as well as during meiosis because of the severe meiotic phenotype of chk-2 loss of function mutants (MacQueen and Villeneuve, 2001).
BRCA1 (breast cancer) mutations are found in 45% of all familial breast cancers (Yoshida and Miki, 2004). The BRCA1 gene performs a multitude of functions in response to DNA damage, most of which are poorly understood. The C. elegans ortholog, brc-1, was identified by yeast 2-hybrid interaction of its product with BRD-1, a putative ortholog of BARD1 (Boulton et al., 2004). RNAi depleted brc-1 and brd-1 worms had a high incidence of X-chromosome nondisjunction (Him phenotype) and increased germ cell death (apoptotic phenotype). In addition, radiation treatment resulted in increased apoptotic cell death, chromosome fragmentation, and radiation hyper-sensitivity, strongly implicating brc-1 in DNA damage response.
Recently, a BRCA2-related gene has been identified and shown to be involved in double-strand break repair by homologous recombination like its mammalian counterpart. Martin et al. (2005) demonstrated that the defect in HR is due to inefficient RAD-51 nuclear localization and a failure to recruit RAD-51 to DSBs.
C. elegans was the first organism in which a mutant phenotype was observed resulting from the loss of a G-tract interacting helicase, implicated in repair. The dog-1 (deletion of G tracts) gene encodes a putative helicase required to maintain homopolymeric dC/dG tracts (Cheung et al., 2002). In humans, loss of function of the orthologous BRCA1-interacting protein BACH1 results in breast cancer susceptibility (Cantor et al., 2001; Cantor et al., 2004). Whereas in mouse, loss of function of a related gene, Rtel (regulator of telomere length) showed telomere loss and displayed many chromosome breaks and fusions (Ding et al., 2004).
In humans, Bloom's syndrome is an autosomal-recessive human disorder caused by mutations in the BLM RecQ helicase and is associated with loss of genomic integrity and an increased incidence of cancer. The C. elegans homolog most similar to the BLM helicase is him-6, which when mutated results in enhanced irradiation sensitivity, a partially defective S-phase checkpoint, and reduced levels of DNA-damage induced apoptosis (Wicky et al., 2004). him-6 and top-3 are needed to prevent the accumulation of double-strand breaks in normally proliferating germ cells, and act in partially redundant pathways downstream of rad-51.
A third helicase, the C. elegans Werner's Syndrome RecQ helicase, WRN-1, is implicated in the DNA damage checkpoint (Li et al., 2004; Lee et al., 2004). Lee et al. (2004) showed that WRN-1 defects are accentuated by gamma-irradiation, implying that they are derived from spontaneous or induced DNA damage. Irrespective of gamma-irradiation, pre-meiotic germ cells had an abnormal checkpoint response to DNA replication blockage. These observations suggest that WRN-1 acts as a checkpoint protein for DNA damage and replication blockage. The authors point out that the wrn-1(RNAi) phenotypes are similar to those of Werner syndrome; for example, premature aging and reduced body size, suggesting that C. elegans may be a useful model for this human disease.
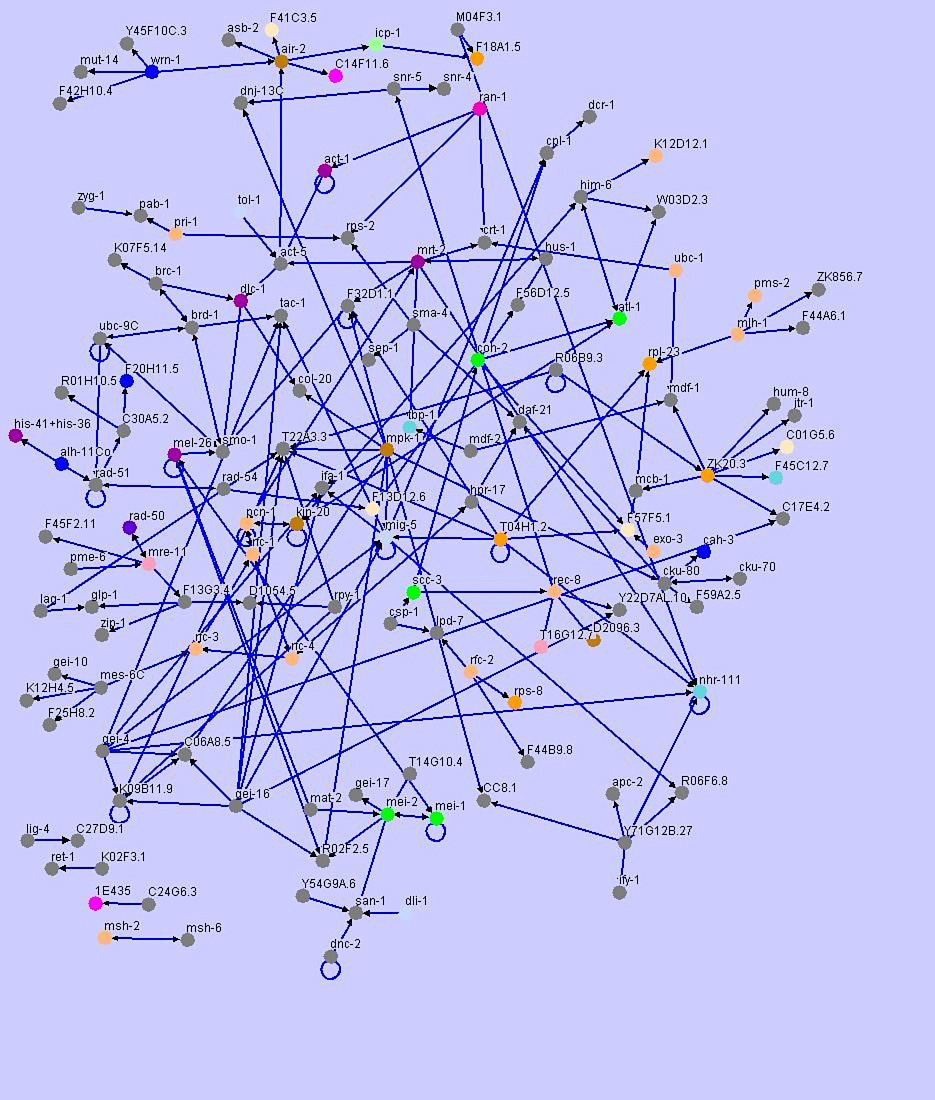
A broad perspective on genes involved in DNA repair has been gained using high throughput, genome-wide analysis of RNAi phenotypes in C. elegans (Piano et al., 2002 Kamath et al., 2003 Pothof et al., 2003; Lettre et al., 2004; van Haaften et al., 2004). As part of a large scale analysis of protein-protein interactions, known proteins implicated in replication, nucleotide excision repair, mismatch repair, base excision repair, non-homologous end joining, homologous recombination and checkpoint pathways were used in yeast 2-hybrid experiments to identify physical interactors in the predicted proteome (Boulton et al., 2002; Li et al., 2004). Results from this study illustrate how sensors, transducers and mediators are shared when generating different responses like chromatin remodeling, altered gene expression and DNA replication (Figure 2). In a striking way, data from C. elegans demonstrates that many of the pathways are interrelated, and that pathway components exhibit previously unrecognized links between repair mechanisms and checkpoints.
|
Figure 2. Yeast 2-hybrid data from Li et al. (2004) showing interactions between DNA damage proteins and chromosomal components. Data has been visualized by Maja Tarailo from the University of British Columbia, using Osprey, a software platform for visualizing complex interaction networks. Colours denote Gene Ontology (GO) terms.
C. elegans has proven to be a useful model with which to investigate the factors maintaining genome stability. Genes have been identified by mutagenesis and RNAi that affect DNA damage checkpoint and repair functions resulting in hypersensitivity to radiation. To date no obvious DNA damage-induced checkpoint has been described in the soma. In contrast, the DNA damage response in the germ line is characterized by two spatially separate checkpoint responses; mitotic germ nuclei proliferation arrest and cep-1-mediated apoptosis of damaged meiotic nuclei. In addition to single gene studies, integration of data from high-throughput screens has identified genes not previously implicated in the DNA damage response and elucidated novel connections between the different repair pathways.
We wish to thank Maja Tarailo, Jill Youds, Peter Lansdorp, David Baillie and our reviewers for thoughtful comments on the chapter; Maja Tarailo for preparation of Figure 2; WormBase and PubMed for resources; and CIHR and NSERC for funding support.
Ahmed, S., and Hodgkin, J. (2000). MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature 403, 159–164. Abstract Article
Ahmed, S., Alpi, A., Hengartner, M.O., and Gartner, A. (2001). C. elegans RAD-5/CLK-2 defines a new DNA damage checkpoint protein. Curr. Biol. 11, 1934–1944. Abstract Article
Aoki, H., Sato, S., Takanami, T., Ishihara, T., Katsura, I., Takahashi, H., Higashitani, A. (2000). Characterization of Ce-atl-1, an ATM-like gene from Caenorhabditis elegans. Mol. Gen. Genet. 264, 119–126. Abstract Article
Alpi, A., Pasierbek, P., Gartner, A., Loidl, J. (2003). Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma 112, 6–16. Abstract Article
Benard, C., McCright, B., Zhang, Y., Felkai, S., Lakowski, B., Hekimi, S. (2001). The C. elegans maternal-effect gene clk-2 is essential for embryonic development, encodes a protein homologous to yeast Tel2p and affects telomere length. Development 128, 4045–4055. Abstract
Bergamaschi, D., Samuels, Y., O'Neil, N.J., Trigiante, G., Crook, T., Hsieh, J.K., O'Connor, D.J., Zhong, S., Campargue, I., Tomlinson, M.L., Kuwabara, P.E., Lu, X. (2003). iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat. Genet. 33, 162–167. Abstract Article
Boulton, S.J., Martin, J.S., Polanowska, J., Hill, D.E., Gartner, A., Vidal, M. (2004). BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr. Biol. 14, 33–39. Abstract Article
Boulton, S.J., Gartner, A., Reboul, J., Vaglio, P., Dyson, N., Hill, D.E., Vidal, M. (2002). Combined functional genomic maps of the C. elegans DNA damage response. Science 295, 127–131. Abstract Article
Buermeyer, A.B., Deschenes, S.M., Baker, S.M., Liskay, R.M. (1999). Mammalian DNA mismatch repair. Annu. Rev. Genet. 33, 533–564. Abstract Article
Cantor, S.B., Bell, D.W., Ganesan, S., Kass, E.M., Drapkin, R., Grossman, S., Wahrer, D.C., Sgroi, D.C., Lane, W.S., Haber, D.A., Livingston, D.M. (2001). BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105, 149–160. Abstract Article
Cantor, S., Drapkin, R., Zhang, F., Lin, Y., Han, J., Pamidi, S., Livingston, D.M. (2004). The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc. Natl. Acad. Sci. USA 101, 2357–2362. (Erratum in: Proc. Natl. Acad. Sci. USA 101, 6834.) Abstract Article
Cheung, I., Schertzer, M., Rose, A.M., Lansdorp, P.M. (2002). Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nature Genetics 31, 405–409. Abstract
Cheung, I., Schertzer, M., Baross, A., Rose, A.M., Lansdorp, P.M., Baird, D.M. (2004). Strain-specific telomere length revealed by single telomere length analysis in Caenorhabditis elegans. Nucleic Acids Res. 32, 3383–3391. Abstract Article
Cheung, I., Schertzer, M., Rose, A.M., Lansdorp, P.M. (in press). High incidence of rapid telomere loss in telomerase-deficient Caenorhabditis elegans. Nucleic Acids Res., in press.
Chin, G.M., Villeneuve, A.M. (2001). C. elegans mre-11 is required for meiotic recombination and DNA repair but is dispensable for the meiotic G(2) DNA damage checkpoint. Genes Dev. 15, 522–534. Abstract Article
Degtyareva, N.P., Greenwell, P., Hofmann, E.R., Hengartner, M.O., Zhang, L., Culotti, J.G., Petes, T.D. (2002). Caenorhabditis elegans DNA mismatch repair gene msh-2 is required for microsatellite stability and maintenance of genome integrity. Proc. Natl. Acad. Sci. USA 99, 2158–2163. Abstract Article
Deng, X., Hofmann, E.R., Villanueva, A., Hobert, O., Capodieci, P., Veach, D.R., Yin, X., Campodonico, L., Glekas, A., Cordon-Cardo, C., Clarkson, B., Bornmann, W.G., Fuks, Z., Hengartner, M.O., Kolesnick, R. (2004). Caenorhabditis elegans ABL-1 antagonizes p53-mediated germline apoptosis after ionizing irradiation. Nat. Genet. 36, 906–912. Abstract Article
Derry, W.B., Putzke, A.P., Rothman, J.H. (2001). Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294, 591–595. Abstract Article
Ding, H., Schertzer, M., Wu, X., Gertsenstein, M., Selig, S., Kammori, M., Pourvali, R., Poon, S., Vulto, I., Chavez, E., Tam, P.P., Nagy, A., Lansdorp, P.M. (2004). Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell 117, 873–886. Abstract Article
Gartner, A., Milstein, S., Ahmed, S., Hodgkin, J., Hengartner, M.O. (2000). A conserved checkpoint pathway mediates DNA damage--induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 5, 435–443. Abstract Article
Hartman, P.S, Herman, R.K. (1982). Radiation-sensitive mutants of Caenorhabditis elegans. Genetics 102, 159–178. Abstract
Hartman, P.S. (1984). UV irradiation of wild type and radiation-sensitive mutants of the nematode Caenorhabditis elegans: fertilities, survival, and parental effects. Photochem. Photobiol. 39(2), 169–175. Abstract
Hartman, P.S. (1984). Effects of age and liquid holding on the UV-radiation sensitivities of wild-type and mutant Caenorhabditis elegans dauer larvae. Mutat. Res. 132(3–4), 95–99. Abstract
Hofmann, E.R., Milstein, S., Boulton, S.J., Ye, M., Hofmann, J.J., Stergiou, L., Gartner, A., Vidal, M., Hengartner, M.O. (2002). Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr. Biol. 12, 1908–1918. Abstract Article
Huyen, Y., Jeffrey, P.D. Derry, W.B., Rothman, J.H., Pavletich, N.P., Stavridi, E.S., Halazonetis, T.D. (2004). Structural differences in the DNA binding domains of human. Abstract Article
Jiang, N., Benard, C.Y., Kebir, H., Shoubridge, E.A., Hekimi, S. (2003). Human CLK2 links cell cycle progression, apoptosis, and telomere length regulation. J. Biol. Chem. 278, 21678–21684. Abstract Article
Jones, C.A., Hartman, P.S. (1996). Replication in UV-irradiated Caenorhabditis elegans embryos. Photochem Photobiol. 63(2), 187–192. Abstract
Kamath, R.S., Fraser, A.G., Dong, Y., Poulin, G., Durbin, R., Gotta, M., Kanapin, A., Le Bot, N., Moreno, S., Sohrmann, M., Welchman, D.P., Zipperlen, P., Ahringer, J. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. Abstract Article
Krogh, B.O., Symington, L.S. (2004) Recombination Proteins in Yeast. Abstract
Lee, M.H., Ahn, B., Choi, I.S., Koo, H.S. (2002). The gene expression and deficiency phenotypes of Cockayne syndrome B protein in Caenorhabditis elegans. FEBS Lett. 522(1–3), 47–51. Abstract Article
Lee, S.J., Yook, J.S., Han, S.M., Koo, H.S. (2004). A Werner syndrome protein homolog affects C. elegans development, growth rate, life span and sensitivity to DNA damage by acting at a DNA damage checkpoint. Development 131(11), 2565–2575. Abstract Article
Lettre, G., Kritikou, E.A., Jaeggi, M., Calixto, A., Fraser, A.G., Kamath, R.S., Ahringer, J., Hengartner, M.O. (2004). Genome-wide RNAi identifies p53-dependent and -independent regulators of germ cell apoptosis in C. elegans. Cell Death Differ. Jul 23 [Epub ahead of print]. Abstract Article
Li, S., Armstrong, C.M., Bertin, N., Ge, H., Milstein, S., Boxem, M., Vidalain, P.O., Han, J.D., Chesneau, A., Hao, T., Goldberg, D.S., Li, N., Martinez, M., Rual, J.F., Lamesch, P., Xu, L., Tewari, M., Wong, S.L., Zhang, L.V., Berriz, G.F., Jacotot, L., Vaglio, P., Reboul, J., Hirozane-Kishikawa, T., Li, Q., Gabel, H.W., Elewa, A., Baumgartner, B., Rose, D.J., Yu, H., Bosak, S., Sequerra, R., Fraser, A., Mango, S.E., Saxton, W.M., Strome, S., Van Den Heuvel, S., Piano, F., Vandenhaute, J., Sardet, C., Gerstein, M., Doucette-Stamm, L., Gunsalus, K.C., Harper, J.W., Cusick, M.E., Roth, F.P., Hill, D.E., Vidal, M. (2004). A map of the interactome network of the metazoan C. elegans. Science 303, 540–543. Abstract Article
Lim, C.S., Mian, I.S., Dernburg, A.F., Campisi, J. (2001). C. elegans clk-2, a gene that limits life span, encodes a telomere length regulator similar to yeast telomere binding protein Tel2p. Curr. Biol. 11, 1706–1710. Abstract Article
MacQueen, A.J., Villeneuve, A.M. (2001). Nuclear reorganization and homologous chromosome pairing during meiotic prophase require C. elegans chk-2. Genes Dev. 15, 1674–1687. Abstract Article
Martin, J.S., Winkelmann, N., Petalcorin, M.I.R., McIlwraith, M.J., Boulton, S.J. (2005). RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol. Cell. Biol. 25, 3127–3139. Abstract Article
Park, H.K, Yook, J.S., Koo, H.S., Choi, I.S., Ahn, B. (2002). The Caenorhabditis elegans XPA homolog of human XPA. Mol. Cells 14, 50–55. Abstract
Park, H.K., Suh, D., Hyun, M., Koo, H.S., Ahn, B. (2004). A DNA repair gene of Caenorhabditis elegans: a homolog of human XPF. DNA Repair(Amst). 3, 1375–1383. Abstract Article
Piano, F., Schetter, A.J., Morton, D.G., Gunsalus, K.C., Reinke, V., Kim, S.K., Kemphues, K.J. (2002). Gene clustering based on RNAi phenotypes of ovary-enriched genes in C. elegans. Curr. Biol. 12, 1959–1964. Abstract Article
Pothof, J., van Haaften, G., Thijssen, K., Kamath, R.S., Fraser, A.G., Ahringer, J., Plasterk, R.H., Tijsterman, M. (2003). Identification of genes that protect the C. elegans genome against mutations by genome-wide RNAi. Genes Dev. 17, 443–448. Abstract Article
Rinaldo, C., Bazzicalupo, P., Ederle, S., Hilliard, M., La Volpe, A. (2002). Roles for Caenorhabditis elegans rad-51 in meiosis and in resistance to ionizing radiation during development. Genetics 160, 471–479. Abstract
Sancar, A., Lindsey-Boltz, L.A., Unsal-Kaccmaz, K., Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 73, 39–85. Abstract Article
Schumacher, B., Hofmann, K., Boulton, S., Gartner, A. (2001). The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 11, 1722–1727. Abstract Article
Shinohara, A, Ogawa, T. (1995). Homologous recombination and the roles of double-strand breaks. Trends Biochem. Sci. 20(10), 387–391. Abstract Article
Stergiou, L., Hengartner, M.O. (2004). Death and more: DNA damage response pathways in the nematode C. elegans. Cell Death Differ. 11, 21–28. Abstract Article
Takanami, T., Sato, S., Ishihara, T., Katsura, I., Takahashi, H., Higashitani, A. (1998). Characterization of a Caenorhabditis elegans recA-like gene Ce-rdh-1 involved in meiotic recombination. DNA Res. 5, 373–377. Abstract Article
Takanami, T., Mori, A., Takahashi, H., Horiuchi, S., Higashitani, A. (2003). Caenorhabditis elegans Ce-rdh-1/rad-51 functions after double-strand break formation of meiotic recombination. Chromosome Res. 11, 125–135. Abstract Article
Takanami, T., Mori, A., Takahashi, H., Higashitani, A. (2000). Hyper-resistance of meiotic cells to radiation due to a strong expression of a single recA-like gene in Caenorhabditis elegans. Nucleic Acids Res. 28, 4232–4236. Abstract Article
Tijsterman, M., Pothof, J., Plasterk, R.H. (2002). Frequent germline mutations and somatic repeat instability in DNA mismatch-repair-deficient Caenorhabditis elegans. Genetics 161, 651–660. Abstract
van Haaften, G., Vastenhouw, N.L., Nollen, E.A., Plasterk, R.H., Tijsterman, M. (2004). Gene interactions in the DNA damage-response pathway identified by genome-wide RNA-interference analysis of synthetic lethality. Proc. Natl. Acad. Sci. USA 101, 12992–12996. Abstract Article
*Edited by Thomas Blumenthal. Last revised March 4, 2005. Published January 13, 2006. This chapter should be cited as: O'Neil, N. and Rose, A. DNA repair (January 13, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.54.1, http://www.wormbook.org.
Copyright: © 2006 Nigel O'Neil and Ann Rose. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: arose@gene.nce.ubc.ca
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.